|
|
| (同じ利用者による、間の31版が非表示) |
| 1行目: |
1行目: |
| <div align="right"> | | <div align="right"> |
| <font size="+1">[https://researchmap.jp/a_yokoseki 横関明男]</font><br> | | <font size="+1">[http://researchmap.jp/read0113019 西澤 正豊]</font><br> |
| ''新潟大学大学院医歯学総合研究科 臓器連関学寄附講座''<br>
| | ''新潟大学 脳研究所 ''<br> |
| <font size="+1">他田正義</font><br>
| | DOI:<selfdoi /> 原稿受付日:2016年1月30日 原稿完成日:2016年月日<br> |
| ''新潟市民病院 脳神経内科''<br>
| | 担当編集委員:[http://researchmap.jp/read0141446 漆谷 真](滋賀医科大学 医学部 神経内科)<br> |
| <font size="+1">[https://researchmap.jp/onoderao 小野寺理]</font><br>
| |
| ''新潟大学脳研究所 脳神経内科学分野''<br> | |
| DOI:<selfdoi /> 原稿受付日:2021年1月29日 原稿完成日:2021年2月18日<br> | |
| 担当編集委員:[http://researchmap.jp/read0141446 漆谷 真](滋賀医科大学 医学部 脳神経内科)<br> | |
| </div> | | </div> |
|
| |
|
| 英:spinocerebellar degeneration 独:spinozerebellärer Degeneration 仏:dégénérescence spinocérébelleuse
| | 英語名:spinocerebellar degeneration |
|
| |
|
| 英語略:SCD | | 英語略:SCD |
|
| |
|
| {{box|text= 脊髄小脳変性症は、小脳や脊髄の系統変性に伴う運動失調症の総称である。脊髄小脳変性症は、弧発性と遺伝性があり、遺伝性は顕性 (優性) 遺伝と潜在 (劣性) 遺伝に分類される。弧発性では、多系統萎縮症が最も頻度が高く、遺伝性は国により頻度が異なり、日本ではMachado-Joseph病 (MJD) 、spinocerebellar ataxia 6 (SCA6) 、spinocerebellar ataxia 31 (SCA31) の頻度が高い。臨床症状は、小脳および小脳への出入力に関する神経障害により、眼球運動障害、構音障害、歩行障害、体幹失調、筋トーヌス低下など様々な症状を認める。脊髄小脳変性症の治療は、失調症状の改善目的にTRH (thyrotropin releasing hormone) 製剤が使用されているが、効果は限定的であり、現在多くの薬剤で治験が実施されている。}}
| | ==概念== |
| | 脊髄小脳変性症は、[[小脳]]あるいはその連絡線維の変性により、主な症状として[[小脳性運動失調]]を呈する疾患の総称である。 |
|
| |
|
| ==背景==
| | 脊髄小脳変性症は従来、神経病理学的所見に基づいて、主に[[脊髄]]を障害するもの、脊髄と小脳を障害するもの、主に小脳を障害するものの3群に分類されてきた。しかし、最近では遺伝形式と臨床症候に基づく簡便な分類が用いられ、脊髄小脳変性症はまず孤発性と遺伝性に大別される。全体の約3分の2を占める孤発性群はさらに、変性が小脳に限局する[[皮質性小脳萎縮症]]([[cortical cerebellar atrophy]]:[[CCA]])と、変性が小脳系だけでなく、[[大脳基底核系]]や[[自律神経系]]、[[錐体路]]にも拡がる[[多系統萎縮症]]([[multiple system atrophy]]:[[MSA]])に分けられる。孤発性群では、多系統萎縮症が約3分の2、皮質性小脳萎縮症が約3分の1を占める。全体の残り3分の1は遺伝性群で、遺伝形式によって優性遺伝性と劣性遺伝性に分けられる。優性遺伝性が9割以上を占める。 |
| 脊髄小脳変性症の報告は、1860年代に[[w:Nikolaus Friedreich|Friedreich]]が,幼小児期に家族性に発症した失調性疾患 ([[Friedreich失調症]]) を報告し、進行期の[[梅毒]]に合併する[[脊髄癆]]と異なる疾患として記載されたことが最初である<ref name=中西孝雄1978>'''中西孝雄 (1978).'''<br>日本における難病研究の現況脊髄小脳変性症. 内科. 41, 191-194 [https://search.jamas.or.jp/link/ui/1978104684 医中誌Web]</ref> 。この報告以降,弧発性、家族性など種々の脊髄小脳変性症が報告され、知見が蓄積されてきた。その一方、臨床所見、病理所見で明確に区別することが困難であったことから、Holmes (1907) 、Greenfield (1954、1958) 、EscourolleおよびMasson (1967)、Skre (1972) 、高橋昭 (1974) 、Oppenheimer (1976) と様々な病型分類が報告されてきた<ref name=中村晴臣1977>'''中村晴臣 (1977).'''<br>脊髄小脳変性症の分類とその主要症状. 神経研究の進歩. 21, 5-13 [http://search.jamas.or.jp/link/ui/1977107283 医中誌Web]</ref> 。
| |
|
| |
|
| 1990年代に入り、[[免疫組織化学|免疫組織]]診断の発達により、弧発性の脊髄小脳変性症の中で最も頻度の高い[[オリーブ橋小脳変性症]] ([[olivopontocerebellar atrophy]]、[[OPCA]]) 、[[パーキンソン病|パーキンソン症状]]を主体とする[[線条体黒質変性症]] ([[striatenigral degeneration]]、[[SND]]) 、および[[自律神経]]症状が主体である[[シャイ・ドレーガー症候群]] ([[Shy-Drager syndrome]]、SDS) では、いずれも残存する[[オリゴデンドログリア]]内に[[αシヌクレイン]]陽性の[[封入体]]を形成することから、同一の疾患であることが明らかとなり<ref name=Wakabayashi1998><pubmed>9682846</pubmed></ref> 、この疾患群は現在では[[多系統萎縮症]] ([[multiple system atrophy]]、[[MSA]]) と診断されるようになった。
| | 遺伝性脊髄小脳変性症の原因遺伝子の同定が進み、分子病態が解明されつつある現状から、脊髄小脳変性症を病理学的な概念である「変性症」に限定せず、運動失調(ataxia)を呈する疾患群として捉えようとする立場や、分子病態に基づいて分類し直そうとする試みがある。 |
|
| |
|
| 同じく1990~2000年代には、分子遺伝学の発達により、遺伝性脊髄小脳変性症の遺伝子座の同定、さらには原因遺伝子の同定が相次いで報告された。今後も次世代シークエンサーなど遺伝子解析手法の発達により、新たな疾患とその原因遺伝子の同定が進むことが予想される。
| | ==多系統萎縮症== |
| | ===概念=== |
| | [[多系統萎縮症]](Multiple system atrophy : MSA)の多系統変性は、小脳系、大脳基底核系、自律神経系の3系統を中心とし、錐体路にも及ぶ。小脳系の系統変性を主体とする病型は、従来、[[オリーブ橋小脳萎縮症]](olivopontoserebellar atrophy:OPCA)、大脳基底核系では[[線条体黒質変性症]](striatonigral degeneration:SND)、自律神経系では[[Shy-Drager症候群]](Shy-Drager syndrome:SDS)と呼ばれてきた。 |
|
| |
|
| | オリーブ橋小脳萎縮症はDejerineとAndré-Thomasによる1900年の報告に始まるが、オリーブ小脳系を超えた病変も認められていた。1964年にAdamsが提唱した線条体黒質変性症においても、黒質線条体だけでなく、オリーブ小脳系の変性を伴うと記載されていた。Shy-Drager症候群はShyとDragerにより1960年に報告されたが、1967年のSchwarzによる4剖検例では、自律神経系を超えた変性が認められていた。こうした経緯から、GrahamとOppenheimerは1969年、病変分布の共通性から、オリーブ橋小脳萎縮症、線条体黒質変性症、Shy-Drager症候群を包括する多系統委縮症という名称を提案した。高橋によるShy-Drager症候群のわが国初の詳細な剖検報告(1969年)でも、Shy-Drager症候群とオリーブ橋小脳萎縮症病変の共通性が指摘されている。 |
|
| |
|
| == 診断 ==
| | その後、多系統萎縮症に共通する疾患特異的バイオマーカーとして、脳幹の[[オリゴデンドログリア]]や神経細胞の細胞質内に特徴的な封入体([[glial cytoplasmic inclusion]]:[[GCI]]、[[neuronal cytoplasmic inclusion]]:[[NCI]])が見出され、多系統萎縮症は疾患単位として確立された。さらに、glial cytoplasmic inclusion、neuronal cytoplasmic inclusionの主な構成成分は、リン酸化された[[α-シヌクレイン]]であることが明らかにされた。 |
| 「運動失調症の医療基盤に関する研究班」の診断基準が用いられている('''表1'''、'''2''')。
| |
|
| |
|
| {| class="wikitable"
| | 多系統萎縮症の診断には、1999年に発表されたGilmanらによるconsensus statementが広く用いられてきた。これによると、多系統萎縮症は診断の確かさによりdefinite、probable、possibleの3群に分類され、さらにオリーブ橋小脳萎縮症も線条体黒質変性症もいずれは自律神経症状を合併することからShy-Drager症候群を除外して、小脳症状と自律神経障害を呈して従来のオリーブ橋小脳萎縮症に相当する多系統萎縮症を[[MSA-C]]、[[パーキンソン症]]状と自律神経障害を呈して従来の線条体黒質変性症に相当する多系統萎縮症を[[MSA-P]]として、多系統萎縮症を臨床的に2分した。2008年には、改訂版が発表され、probableとpossibleの主な分岐点は、自律神経症状の程度により規定された。排尿障害では[[尿失禁]]、男性では[[勃起障害]]が重視され、起立性低血圧では、起立後3分以内に収縮期血圧が30 mmHg以上,あるいは拡張期血圧が15 mmHg以上低下する場合をprobableとする基準値が定められた(表1)。 |
| |+表1. 脊髄小脳変性症の診断基準(表2のDefinite、Probableを対象とする)
| | |
| |-
| | これに対してわが国では、[[MSA-A]]としてShy-Drager症候群を残そうとする立場もある。新潟大学脳研究所で、病理学的に診断が確定された多系統萎縮症の臨床像を検討すると、MCA-C、MSA-Pのいずれも22%は、初発症状が自律神経障害であった。Shy-Drager症候群とされてきた症例は、早期から著明な自律神経障害で発症し、次第に小脳性運動失調やパーキンソン症状を伴うが、Shy-Drager症候群に特異的な自律神経障害は指摘できない。また「premotor MSA」(発症早期に自律神経障害が前景に立ち、他の系統変性による症候がまだ目立たない段階で、たまたま病理学的検索が行われた症例)では、オリーブ橋小脳系と線条体黒質系の変性は軽微であるのに対し、脳幹の自律神経諸核には既にglial cytoplasmic inclusionを認めている。また、Shy-Drager症候群と[[進行性自律神経機能不全症]](progressive autonomic failure:PAF)との鑑別も、初期には困難である。こうした知見を総合すると、Shy-Drager症候群を独立した疾患とすることは現時点では難しいと考えられる。 |
| ! style="text-align:left;"|主要項目
| | |
| |-
| | MSA-CとMSA-Pの頻度には、著明な人種差がある。わが国ではMSA-Cが全体の7、8割、MSA-Pが2、3割を占めるが、欧米ではこの頻度が逆転している。MSA-CとMSA-Pは臨床診断であるが、病理学的に診断が確定されたdefinite 多系統萎縮症についても、Wenningらが検討した欧州ではMSA-Pが8割を占め、一方、新潟大学の多系統萎縮症連続剖検例では、MSA-Cが3分の2を占めた。 |
| | 脊髄小脳変性症は、[[運動失調]]を主要症候とする[[神経変性疾患]]の総称であり、臨床、病理あるいは遺伝子的に異なるいくつかの病型が含まれる。臨床的には以下の特徴を有する。<br>
| | |
|
| | 病理所見としては、MSA-Cでは小脳皮質、[[橋]]小脳系、および[[下オリーブ核]]に強い変性と神経細胞脱落、[[グリオーシス]]が認められる。一方、MSA-Pでは[[被殻]]、黒質の変性が高度であり、特に被殻の後外側部は神経細胞脱落が強く、褐色調の色素沈着がみられる。Shy-Drager症候群とされた剖検例では、脊髄中間外側核、迷走神経背側核、交感神経節などの自律神経諸核の変性が強い。 |
| ① 小脳性ないしは[[後索性]]の運動失調又は[[痙性対麻痺]]を主要症候とする。<br>
| | |
| ② 徐々に発病し、経過は緩徐進行性である。<br>
| | glial cytoplasmic inclusionの主要な構成タンパク質であるα-シヌクレインは、もともとオリゴデンドログリアには発現していない。多系統萎縮症では病的グリアがα-シヌクレインを産生するという可能性よりも、神経細胞が産生したα-シヌクレインが細胞間を伝搬してグリアに取り込まれるという「[[プリオン]]様のタンパク伝搬仮説」が現在は有力である。パーキンソン病の特徴である[[レヴィー小体]]の主な構成成分もリン酸化α-シヌクレインであるが、同じシヌクレイノパチーである多系統萎縮症とパーキンソン病がどこで分岐するかは未解明である。α-シヌクレイン遺伝子の点変異は家族性パーキンソン病の原因とはなるが、多系統萎縮症の表現型は示さない。α-シヌクレイン遺伝子のduplication、あるいはtriplicationによるまれな家族性パーキンソン病では、レヴィー小体とglial cytoplasmic inclusionがともに認められることから、遺伝子量の増大はglial cytoplasmic inclusion形成の原因の一つと考えられる。 |
| ③ 病型によっては遺伝性を示す。その場合、[[常染色体優性遺伝性]]であることが多いが、常染色体あるいは[[X染色体劣性遺伝性]]の場合もある。<br>
| | |
| ④ その他の症候として、[[錐体路症候]]、[[パーキンソニズム]]([[振戦]]、[[筋強剛]]、[[無動]])、[[自律神経]]症候([[排尿]]困難、[[発汗]]障害、[[起立性低血圧]])、[[末梢神経]]症候([[しびれ感]]、[[表在感覚]]低下、[[深部覚]]低下)、[[高次脳機能障害]]([[幻覚]][非薬剤性]、[[失語]]、[[失認]]、[[失行]][肢節運動失行以外])などを示すものがある。<br>
| | ごくまれではあるが、多系統萎縮症には家族発症例があり、これらの解析から辻らにより[[COQ2]]([[コエンザイムQ10合成酵素]])遺伝子に変異が同定された。変異が2つあれば発症者となり、変異が1つでは発症リスクを高めることになる。日本人のみに認められるV393A変異は多系統萎縮症の約9%に見出され(健常者では約3%)、ホモ変異例では脳内のコエンザイムQ10量が減少していた。 |
| ⑤ 頭部[[MRI]]やX線[[CT]]にて、小脳や[[脳幹]]の萎縮を認めることが多いが、病型や時期によっては[[大脳基底核]]病変や[[大脳皮質]]の萎縮などを認めることもある。<br>
| |
| ⑥ 以下の原因による二次性小脳失調症を鑑別する:[[脳血管障害]]、[[腫瘍]]、[[アルコール]]中毒、[[ビタミンB1]]・[[ビタミンB12|B12]]・[[葉酸]]欠乏、薬剤性([[フェニトイン]]など)、炎症[[神経梅毒]]、[[多発性硬化症]]、[[傍腫瘍性神経症候群#小脳変性症|傍腫瘍性小脳炎]]、[[免疫介在性小脳炎]]([[橋本脳症]]、[[シェーグレン症候群]]、[[グルテン失調症]]、[[抗GAD (glutamic acid decarboxylase)抗体小脳炎]]))、[[甲状腺機能低下症]]、[[低セルロプラスミン血症]]、[[脳腱黄色腫症]]、[[ミトコンドリア病]]、[[二次性痙性対麻痺]]([[脊柱疾患]]に伴う[[ミエロパチー]]、脊髄の占拠性病変に伴うミエロパチー、多発性硬化症、[[視神経脊髄炎]]、[[脊髄炎]]、[[HTLV-I (Human T-cell leukemia virus type 1) 関連ミエロパチー]]、[[アルコール性ミエロパチー]]、[[副腎ミエロニューロパチー]]など。<br>
| |
| |}
| |
|
| |
|
| {| class="wikitable" | | {| class="wikitable" |
| |+表2. 診断のカテゴリー | | |+ 表1.多系統萎縮症診断基準改訂版<ref><pubmed>18725592</pubmed></ref> |
| |- | | |- |
| ! style="text-align:left;"|Definite
| | |'''従来通り、definite, probable, possibleに分類し、さらにMSA-PとMSA-Cに分類する。''' |
| | #Definite MSA<br> 病理学的に,中枢神経に広範に、多数のα-synuclein陽性glial cytoplasmic inclusion(GCI)を認め、線条体黒質系またはオリーブ橋小脳系の変性所見を伴う。 |
| | #Probable MSA<br> 孤発性で進行性の成人発症(30歳以降)の変性疾患で、自律神経障害(尿失禁(膀胱からの尿排出をコントロールできない、男性では勃起障害)、または起立後3分以内に少なくとも収縮期血圧が30 mmHg,拡張期血圧が15 mmHg低下する起立性低血圧)に加え、レボドパ反応性の乏しいパーキンソニズム(動作緩慢に、筋強剛、振戦、または姿勢反射障害を伴う)、または小脳症候群(歩行失調に、小脳性構音障害、四肢失調、または小脳性眼球運動障害を伴う)を呈する。 |
| | #Possible MSA<br> 孤発性で進行性の成人発症(30歳以降)の変性疾患で、パーキンソニズム、または小脳症候群を呈し、加えて自律神経障害を示唆する所見(他の原因では説明できない尿意促迫、頻尿、残尿、男性では勃起不全、またはprobable MSAの規準を満たさないレベルの起立性低血圧)を少なくとも一つ認め、さらに以下の表で少なくとも一つの所見を満たすもの。<br> |
| | (1) Possible MSA-P またはMSA-C<br> 腱反射亢進を伴うBabinski徴候陽性、喘鳴。<br> |
| | (2) Possible MSA-P<br> 急速進行性のパーキンソニズム、レボドパ反応性が乏しいこと、運動症状出現3年以内の姿勢反射障害、 |
| | 歩行失調・小脳性構音障害・四肢失調・または<br> 小脳性眼球運動障害、運動症状出現5年以内の嚥下障害、 |
| | MRIにおける被殻・中小脳脚・橋・または小脳の萎縮、FDG-PETにおける被殻・脳幹・または小脳の低代謝。<br> |
| | (3) Possible MSA-C<br> パーキンソニズム(動作緩慢と筋強剛)、MRIにおける被殻・中小脳脚・または橋の萎縮、FDG-PETにおける被殻の低代謝、SPECTまたはPETにおける<br> 黒質線条体ドーパミン作動性ニューロンの節前性脱神経 。 |
| |- | | |- |
| | 脊髄小脳変性症・痙性対麻痺に合致する症候と経過があり、遺伝子診断か神経病理学的診断がなされている場合。 | | |'''多系統萎縮症の診断を支持するred flag所見<br>''' |
| | 口部顔面ジストニア、頸部前屈、カンプトコルミア(脊柱の高度の前屈)and/or Pisa症候群(脊柱の高度の側屈)、手または足の拘縮、吸気時のため息、高度の発声困難、高度の構音障害、いびきの出現または増悪、手足の冷感、病的笑いまたは病的泣き、jerkyなミオクローヌス様の姿勢振戦または動作性振戦。 |
| |- | | |- |
| ! style="text-align:left;"|Probable
| | |'''多系統萎縮症の診断を支持しない所見<br>''' |
| | 典型的丸薬丸め様の静止時振戦、臨床的に有意な末梢神経障害、薬剤誘発性でない幻覚、75歳以上の発症、失調症やパーキンソニズムの家族歴、認知症(DSM-IVによる)、多発性硬化症を示唆する大脳白質病変。 |
| |- | | |- |
| | (1)脊髄小脳変性症に合致する症候があり、診断基準の主要項目①②⑤及び⑥を満たす場合、若しくは痙性対麻痺に合致する症候があり、主要項目①②及び⑥を満たす場合。<br>
| |
| 又は<br>
| |
| (2)当該患者本人に脊髄小脳変性症・痙性対麻痺に合致する症状があり、かつその家系内の他の発症者と同一とみなされる場合(遺伝子診断がなされていない場合も含む。)。
| |
| |-
| |
| ! style="text-align:left;"|Possible
| |
| |-
| |
| | 脊髄小脳変性症・痙性対麻痺に合致する症候があり、診断基準の主要項目①②⑤を満たす、又は痙性対麻痺に合致する症候があり、主要項目①②を満たすが、⑥が除外できない場合。
| |
| |} | | |} |
|
| |
|
| 重症度分類:modified Rankin Scale(mRS)、食事・栄養、呼吸のそれぞれの評価スケールを用いて、いずれかが3以上が指定難病の申請基準である。
| | ===症候=== |
| [[ファイル:Yokoseki spinocerebellar degeneration Fig1.png|サムネイル|'''図1 特発性小脳失調症 (IDCA) の頭部MRI画像'''<br>'''(A)''' 70歳女性の正常MRI、T2強調画像水平断。<br>'''(B、C)''' 発症年齢55歳、経過16年(71歳)のIDCA症例の頭部MRI、T2協調画像。(B)T2強調画像水平断、(C) T2強調画像矢状断。SCA1、SCA3、MJD、SCA6、SCA31、DRPLAの原因遺伝子に異常は認めない。構音障害、歩行障害を認めるが、自力歩行はかろうじて可能。小脳の萎縮を認めるが、脳幹は保たれている。]]
| | MSA-Cは40~60歳に、多くは小脳性運動失調から発症し、次第に自律神経症状や錐体外路症状、錐体路症状を伴う。新潟大学の剖検例では、MSA-Cにパーキンソニズムを伴うのは74%であった。また、尿失禁や排尿困難、起立性低血圧や失神、男性では陰萎などの自律神経症状が発現する中央値は発症から2.5年であり、2.5年より早期から自律神経障害が出現すると、その後の進行が速かった。 |
|
| |
|
| == 分類 ==
| | MSA-Pの多くはパーキンソン症状から発症し、次第に自律神経症状を伴う。小脳性運動失調症状はパーキンソン症状にマスクされやすく、MSA-Pが小脳性運動失調を伴う頻度は、新潟大学の検討では44%であった。MSA-Pの初期には、パーキンソン病との鑑別が困難な症例もある。パーキンソン病に比べて、[[レボドパ]]補充療法の効果が乏しく、進行が速く、症状の左右差や静止時振戦がまれであることが特徴とされるが、MSA-Pでもパーキンソン症状の左右差が明らかな例や、典型的な静止時振戦を示す例、レボドパも無効ではなく、改善を示す例がある。進行期になると、多系統萎縮症でも大脳皮質の著明な萎縮や、進行性の認知障害が認められる。 |
| ここでは遺伝の有無に基づく分類をする。
| |
| === 弧発性脊髄小脳変性症 ===
| |
| ==== 多系統萎縮症 ====
| |
| [[多系統萎縮症]]は、[[弧発性脊髄小脳変性症]]で最も頻度が高い。詳細は[[多系統萎縮症]]の項目を参照。
| |
|
| |
|
| ====皮質性小脳萎縮症====
| | 多系統萎縮症の全経過は約9年で、[[wikipedia:ja:誤嚥性肺炎|誤嚥性肺炎]]や[[wikipedia:ja:敗血症|敗血症]]などの[[wikipedia:ja:感染症|感染症]]が死因となることが多いが、夜間の[[突然死]]も重要である。通常の低音のいびきとは異なる高調の喉頭喘鳴は、[[声帯外転麻痺]]を示唆する症候とされ、声帯外転麻痺による気道閉塞が突然死の原因と考えられてきた。しかし、麻酔薬により睡眠状態を再現して喉頭内視鏡検査を行うと、気道狭窄が生じている部位は[[wikipedia:ja:声帯|声帯]]に限らず、[[wikipedia:ja:被裂部|被裂部]]、[[wikipedia:ja:喉頭蓋|喉頭蓋]]、[[wikipedia:ja:舌根部|舌根部]]、[[wikipedia:ja:軟口蓋|軟口蓋]]など広範囲に及び、また吸気時に喉頭蓋が気管に引き込まれ、気道を閉塞する[[wikipedia:floppy epiglottis|floppy epiglottis]]と呼ばれる病態も合併することが明らかになった。MSAの睡眠呼吸障害に対する治療法として、マスクを用いた[[持続陽圧換気]](continuous positive airway pressure: CPAP)を不用意に行うと、floppy epiglottisでは気道狭窄が悪化する恐れがあり、注意を要する。 |
| [[皮質性小脳萎縮症]] ([[cortical cerebellar atrophy]]、[[CCA]]; 純粋小脳型の失調症) は、[[小脳皮質]]の萎縮が主病変とする失調症の総称である ('''図1''') 。多くは高齢発症であることから、[[晩発性皮質性小脳萎縮症]] ([[late cortical cerebellar atrophy]]、[[LCCA]]) とも呼ばれてきた疾患群である。つまり、皮質性小脳萎縮症は単一の疾患ではなく、小脳皮質が比較的選択的に変性、脱落する疾患の一群を示している。
| |
|
| |
|
| 成人期に発症し、緩徐進行性の小脳失調を主体とする。
| | 多系統萎縮症の睡眠呼吸障害に対して、CPAP装着や気管切開などを行っても、突然死を防げない症例が存在する。[[中枢性無呼吸]]や致死性[[wikipedia:ja:不整脈|不整脈]]などが原因と考えられ、気管切開による人工呼吸管理が必要になる。 |
|
| |
|
| これまで[[sporadic adult-onset ataxia of unknown origin]] ([[SAOA]]) <ref name=Abele2007><pubmed>17934884</pubmed></ref> 、[[特発性小脳失調症]]([[idiopathic cerebellar ataxia]], [[IDCA]]) <ref name=Burk2004><pubmed>14570820</pubmed></ref> 、[[idiopathic cerebellar ataxia of late onset]] <ref name=Klockgether1990><pubmed>2341843</pubmed></ref>など報告者によって様々な疾患名で呼ばれてきた経緯があり、疾患概念に混乱が生じていた。その理由は、診断特異的[[バイオマーカー]]や特異的なタンパク質の蓄積などが発見されていないため、除外診断によりなされる点である。つまり皮質性小脳萎縮症の診断においては、初期の多系統萎縮症、[[自己免疫性失調症]]、spinocerebellar ataxia 6 (SCA6)やspinocerebellar ataxia 31 (SCA31) のように小脳失調が主体の遺伝性脊髄小脳変性症など、高齢発症の他の脊髄小脳変性症を除外することが必須である。
| | ===補助診断法=== |
| | [[image:脊髄小脳変性症1.png|thumb|350px|'''図1.多系統萎縮症のMRI所見'''<br>図左:MSA-Cにおける橋十字サインと橋、小脳の萎縮<br> |
| | 図右:MSA-Pにおける線条体後外側部の線状高信号(スリットサイン)]] |
|
| |
|
| このことから、本邦の「運動失調症の医療基盤に関する調査研究班」では、皮質性小脳萎縮症や晩発性皮質性小脳萎縮症に変わる臨床診断名として、[[特発性小脳失調症]]を提唱し、診断基準を策定した<ref name=Yoshida2018><pubmed>29249373</pubmed></ref> 。本邦での特発性小脳失調症と従来報告されてきたSAOAを比較すると、特発性小脳失調症は小脳症状以外の神経症状の合併頻度が少なく、特に[[錐体路症状]]や[[排尿障害]]の合併はSAOAと比べて本邦の特発性小脳失調症は低いことが明かとなっている。つまり、本邦の特発性小脳失調症はより純粋小脳型の失調症を反映していると考えられる<ref name=Yoshida2018><pubmed>29249373</pubmed></ref> 。今後、これらの知見の集積により、特発性小脳失調症から新たな疾患が分離独立することが予想される。
| | 多系統萎縮症の補助診断には[[MRI]]が有用である。MSA-Cでは、小脳、[[中小脳脚]]、[[脳幹]]の進行性萎縮とともに、橋底部に十字状の高信号(hot cross bun sign:橋十字サイン)が、MSA-Pでは、被殻の進行性萎縮とグリオーシス、鉄の沈着により、被殻後外側部に線状の高信号(putaminal slit sign)が認められる(図1)。 |
|
| |
|
| === 遺伝性脊髄小脳変性症 ===
| | [[MIBG心筋シンチグラフィー]]では、MSA-Pの初期には取り込みの低下は認められないので、パーキンソン病との鑑別に役立つ。[[脳脊髄液]]中のα-シヌクレインは多系統萎縮症では低下する。glial cytoplasmic inclusionに結合するリガンドを利用した[[PET]]検査も開発中である。 |
| [[wj:顕性遺伝|顕性 (優性) 遺伝]]と[[wj:潜性遺伝|潜性 (劣性) 遺伝]]様式を取るものが存在する。 | |
|
| |
|
| ==== 顕性遺伝型==== | | ===治療=== |
| 顕性 (優性) 遺伝 を示し、遺伝子座が同定されたものは[[脊髄小脳失調症]] ([[spinocerebellar ataxia]], [[SCA]]) として命名されている。1993年に原因遺伝子が同定された[[spinocerebellar ataxia 1]] ([[SCA1]])から2021年1月の時点で、[[SCA48]]まで同定されている ('''表3''') 。
| | 根治的治療法は確立されておらず、対症療法が主体となる。わが国では、[[thyrotropin releasing hormone]]([[TRH]])の点滴とその誘導体([[タルチレリン]])の経口投与が、小脳性運動失調に対して唯一保険適用となっているが、その効果は限定的である。起立性低血圧や排尿障害などの自律神経症状には、対症療法を行う。多くの薬剤について、小脳性運動失調症に対する有効性が検証されているが、確実に効果が実証されたものはない。 |
|
| |
|
| 原因遺伝子により発症年齢が異なるが、SCA1、[[spinocerebellar ataxia 2]] ([[SCA2]])、[[マチャド・ジョセフ病]] ([[Machado-Joseph disease]], [[MJD]]。[[spinocerebellar ataxia 3]] ([[SCA3]])と同義)、[[spinocerebellar ataxia 6]] ([[SCA6]])、[[歯状核赤核淡蒼球ルイ体萎縮症]] ([[dentatorubral-pallidoluysian atrophy]], [[DRPLA]]) のように遺伝子変異がC(シトシン)、A(アデニン)、G(グアニン)、3塩基の組み合わせである[[CAGリピート]]の異常伸長により発症する疾患は、CAGリピート長が長いほど、発症年齢は若年化し、より重症化する。また親から子に異常遺伝子が伝達される際に、子どものCAGリピート長が親のリピート長より伸長することにより、子供の発症年齢の若年化、症状が重症化する[[表現促進現象]]を認める。ただし、SCA6では、表現促進現象は認められてない<ref name=Ishikawa1997><pubmed>9311738</pubmed></ref> 。
| | 多系統萎縮症では、経過中に気道や尿路の感染症を繰り返して、全身状態が悪化することが多い。口腔ケアを徹底して、誤嚥による気道感染を予防することが重要である。 |
| | |
| {| class="wikitable"
| |
| |+表3. 顕性遺伝の脊髄小脳変性症
| |
| !疾患!!原因遺伝子/遺伝子座!!Phenotype MIM number!!遺伝子変異および臨床的特徴
| |
| |-
| |
| | [[SCA1]] || [https://www.omim.org/entry/601556 ATXN1]|| [https://www.omim.org/entry/164400 164400] || CAGリピート伸長、表現促進現象
| |
| |-
| |
| |[[SCA2]] || [https://www.omim.org/entry/601517 ATXN2]|| [https://www.omim.org/entry/183090 183090] || CAGリピート伸長、表現促進現象、slow eye movement
| |
| |-
| |
| |[[MJD]]/[[SCA3]] || [https://www.omim.org/entry/607047 ATXN3 ]|| [https://www.omim.org/entry/109150 109150 ]|| CAGリピート伸長、表現促進現象、bulging eyes、末梢神経障害
| |
| |-
| |
| | [[SCA4]] || 16q22.1 || [https://www.omim.org/entry/600223 600223 ]|| axonal sensory neuropathy
| |
| |-
| |
| | [[SCA5]] || [https://www.omim.org/entry/604985 SPTBN2 ]|| [https://www.omim.org/entry/600224 600224 ]|| 進行が遅い
| |
| |-
| |
| | [[SCA6]] || [https://www.omim.org/entry/601011 CACNA1A ]|| [https://www.omim.org/entry/183086 183086 ]|| CAGリピート伸長、表現促進現象、発症は50歳以降
| |
| |-
| |
| | [[SCA7]] || [https://www.omim.org/entry/607640 ATXN7 ]|| [https://www.omim.org/entry/164500 164500 ]|| CAGリピート伸長、[[網膜色素変性症]]
| |
| |-
| |
| | [[SCA8]] || [https://www.omim.org/entry/613289 ATXN8 ]|| [https://www.omim.org/entry/608768 608768 ]|| CAGリピート伸長、表現促進現象、[[振動覚]]低下、進行は遅い
| |
| |-
| |
| | [[SCA9]] || - || [https://www.omim.org/entry/612876 612876 ]|| 英国起源の米国人家系、臨床症状はMJDに類似
| |
| |-
| |
| | [[SCA10]] || [https://www.omim.org/entry/611150 ATXN10 ]|| [https://www.omim.org/entry/603516 603516 ]|| ATTCTリピート伸長、[[脳波]]異常、[[てんかん]]
| |
| |-
| |
| | [[SCA11]] || [https://www.omim.org/entry/611695 TTBK2 ]|| [https://www.omim.org/entry/604432 604432 ]|| 症状はSCA6に類似
| |
| |-
| |
| | [[SCA12]] || [https://www.omim.org/entry/604325 PPP2R2B ]|| [https://www.omim.org/entry/604326 604326 ]|| CAGリピート異常伸長(5’UTR)、振戦(上肢、頭部)、[[腱反射]]亢進、[[認知症]]
| |
| |-
| |
| | [[SCA13]] || [https://www.omim.org/entry/176264 KCNC3 ]|| [https://www.omim.org/entry/605259 605259 ]|| [[精神発達遅滞]]
| |
| |-
| |
| | [[SCA14]] || [https://www.omim.org/entry/176980 PRKCG ]|| [https://www.omim.org/entry/605361 605361 ]|| 間欠的な体幹の[[ミオクローヌス]]
| |
| |-
| |
| | [[SCA15]]/[[SCA16|16]] || [https://www.omim.org/entry/147265 ITPR1 ]|| [https://www.omim.org/entry/606658 606658 ]|| 成人発症、進行は遅い
| |
| |-
| |
| | [[SCA17]] || [https://www.omim.org/entry/600075 TBP ]|| [https://www.omim.org/entry/607136 607136 ]|| CAG/CAAリピート伸長、[[認知機能障害]]、[[精神症状]]
| |
| |-
| |
| | [[SCA18]] || 7q22-q23 || [https://www.omim.org/entry/607458 607458 ]|| [[末梢神経障害]]
| |
| |-
| |
| | [[SCA19]]/[[SCA22|22]] || [https://www.omim.org/entry/605411 KCND3 ]|| [https://www.omim.org/entry/607346 607346 ]|| 認知機能障害、ミオクローヌス、振戦
| |
| |-
| |
| | [[SCA20]] || 11q12.2-11q12.3 || [https://www.omim.org/entry/608687 608687 ]|| けいれん性[[発声障害]]、[[喉頭筋]]麻痺
| |
| |-
| |
| | [[SCA21]] || [https://www.omim.org/entry/616101 TMEM240 ]|| [https://www.omim.org/entry/607454 607454 ]|| 認知機能障害、振戦
| |
| |-
| |
| | [[SCA23]] || [https://www.omim.org/entry/131340 PDYN ]|| [https://www.omim.org/entry/610245 610245 ]|| 末梢神経障害
| |
| |-
| |
| | [[SCA24]]<br>→[[SCAR24]] || || ||
| |
| |-
| |
| | [[SCA25]] || 2p15-p21 || [https://www.omim.org/entry/608703 608703 ]|| 末梢性感覚障害、[[視力]]低下、顔面の[[チック]]、頻尿、消化器症状
| |
| |-
| |
| | [[SCA26]] || [https://www.omim.org/entry/130610 EEF2 ]|| [https://www.omim.org/entry/609306 609306 ]|| 純粋小脳型
| |
| |-
| |
| | [[SCA27]] || [https://www.omim.org/entry/601515 FGF14 ]|| [https://www.omim.org/entry/609307 609307 ]|| 振戦、顔面の[[ジスキネジア]]、認知機能障害
| |
| |-
| |
| | [[SCA28]] || [https://www.omim.org/entry/604581 AFG3L2 ]|| [https://www.omim.org/entry/610246 610246 ]|| slow saccades、[[眼筋]]麻痺、[[眼瞼下垂]]
| |
| |-
| |
| | [[SCA29]] || [https://www.omim.org/entry/147265 ITPR1 ]|| [https://www.omim.org/entry/117360 117360 ]|| SCA15と同遺伝子、若年発症、軽度認知機能発達遅滞、非常に緩徐進行
| |
| |-
| |
| | [[SCA30]] || 4q34.3-q35.1. || [https://www.omim.org/entry/613371 613371 ]|| オーストラリア1家系6名、発症年齢平均52歳、腱反射亢進
| |
| |-
| |
| | [[SCA31]] || [https://www.omim.org/entry/612051 BEAN1 ]|| [https://www.omim.org/entry/117210 117210 ]|| イントロンTGGAAリピート伸長、純粋小脳型、[[感音性難聴]]
| |
| |-
| |
| | [[SCA32]] || 7q32-q33 || [https://omim.org/entry/613909 613909 ]|| 中国1家系、認知機能障害、[[無精子症]]
| |
| |-
| |
| | [[SCA33]] || 登録なし || ||
| |
| |-
| |
| | [[SCA34]] || [https://www.omim.org/entry/605512 ELOVL4 ]|| [https://www.omim.org/entry/133190 133190 ]|| [[紅斑]]、[[魚鱗癬]]、[[角質増殖症]]、認知機能は正常
| |
| |-
| |
| | [[SCA35]] || [https://www.omim.org/entry/613900 TGM6 ]|| [https://www.omim.org/entry/613908 613908 ]|| 進行は緩徐、手の振戦
| |
| |-
| |
| | [[SCA36]] || [https://www.omim.org/entry/614154 NOP56 ]|| [https://www.omim.org/entry/614153 614153 ]|| イントロンGGCCTGリピート伸長、舌の線維束性収縮、上位運動ニューロン徴候
| |
| |-
| |
| | [[SCA37]] || [https://www.omim.org/entry/603448 DAB1 ]|| [https://www.omim.org/entry/615945 615945 ]|| 発症年齢は10台後半~60歳代、緩徐進行
| |
| |-
| |
| | [[SCA38]] || [https://www.omim.org/entry/611805 ELOVL5 ]|| [https://www.omim.org/entry/615957 615957 ]|| 末梢神経障害、認知機能は正常
| |
| |-
| |
| | [[SCA39]] || 11q21-q22.3 || 登録なし || フランスの1家系、spastic ataxia
| |
| |-
| |
| | [[SCA40]] || [https://www.omim.org/entry/611204 CCDC88C ]|| [https://www.omim.org/entry/616053 616053 ]|| [[企図振戦]]、[[痙性対麻痺]]、腱反射亢進
| |
| |-
| |
| | [[SCA41]] || [https://www.omim.org/entry/602345 TRPC3 ]|| [https://www.omim.org/entry/616410 616410 ]|| 成人期発症、[[神経伝導速度]]に異常なし
| |
| |-
| |
| | [[SCA42]] || [https://www.omim.org/entry/604065 CACNA1G ]|| [https://www.omim.org/entry/616795 616795 ]|| 発症年齢に大きな差(9~78歳)、振動覚低下や排尿症状(頻度少ない)、進行は比較的緩徐
| |
| |-
| |
| | [[SCA43]] || [https://www.omim.org/entry/120520 MME ]|| [https://www.omim.org/entry/617018 617018 ]|| 末梢神経障害、[[凹足]]、[[下肢]][[筋萎縮]]、認知機能正常
| |
| |-
| |
| | [[SCA44]] || [https://www.omim.org/entry/604473 GRM1 ]|| [https://www.omim.org/entry/617691 617691 ]|| 20-50歳代発症、緩徐進行、車椅子使用までにならず、腱反射は正常~亢進
| |
| |-
| |
| | [[SCA45]] || [https://www.omim.org/entry/604269 FAT2 ]|| [https://www.omim.org/entry/617769 617769 ]|| 40歳以降発症、純粋小脳型
| |
| |-
| |
| | [[SCA46]] || [https://www.omim.org/entry/615698 PLD3 ]|| [https://www.omim.org/entry/617770 617770 ]|| オランダの大家系、成人発症、感覚性ニューロパチー
| |
| |-
| |
| | [[SCA47]] || [https://www.omim.org/entry/607204 PUM1 ]|| [https://www.omim.org/entry/617931 617931 ]|| 若年発症(重症はてんかん性脳症、筋トーヌス低下)、成人発症
| |
| |-
| |
| | [[SCA48]] || [https://www.omim.org/entry/607207 STUB1 ]|| [https://www.omim.org/entry/618093 618093 ]|| 認知機能障害、不安症状、遂行機能障害
| |
| |-
| |
| | [[DRPLA]] || [https://www.omim.org/entry/607462 ATN1 ]|| [https://www.omim.org/entry/125370 125370 ]|| CAGリピート伸長、表現促進現象、ミオクローヌスてんかん
| |
| |}
| |
|
| |
|
| SCA= spinocerebellar ataxia, MJD=[[マチャド・ジョセフ病]], DRPLA=歯状核赤核淡蒼球ルイ体萎縮症 (dentatorubral-pallidoluysian atrophy)
| | 脊髄小脳変性症と多系統萎縮症は[[wikipedia:ja:厚生労働省|厚生労働省]]の[[wikipedia:ja:指定難病制度|指定難病制度]]の対象疾患であり、さらに介護保健法における「特定疾病」に指定されている。制度上Shy-Drager症候群を拡大して多系統萎縮症として独立させたために、脊髄小脳変性症には皮質性小脳萎縮症と遺伝性脊髄小脳変性症が残された形となっている。また、MSA-Pはパーキンソン病と診断されている場合が少なからずあり、難病対策制度上の分類には、再度整理が必要である。 |
|
| |
|
| ==== 潜性遺伝型==== | | ==皮質性小脳萎縮症== |
| [[Friedreich失調症]] ([[Friedreich ataxia]]、[[FRDA]])、[[毛細血管拡張性小脳失調症]] ([[ataxia telangiectasia]]、AT) など一部の疾患を除き、原因遺伝子座が同定されたものは[[常染色体潜性遺伝性脊髄小脳失調症]] ([[spinocerebellar ataxia, autosomal recessive]], [[SCAR]]) と命名され、SCAR28まで同定されている ('''表4''') 。潜性遺伝の脊髄小脳変性症は、一般的に原因遺伝子タンパク質の機能喪失 (loss of function) により発症すると考えられており、そのため若年発症の疾患が多い。
| | ===概念=== |
| | 脊髄小脳変性症の中では最も高齢で発症し、小脳性運動失調のみが緩徐に進行する孤発性の一群を[[皮質性小脳萎縮症]](Cortical cerebellar atrophy : CCA)と呼んでいる。しかし、皮質性小脳萎縮症は単一疾患ではなく、一見家族歴を欠いていても、遺伝子診断により後述する[[SCA6]]や[[SCA31]]と確定される例があり、またアルコール性などの二次性小脳変性症も含まれる。純粋小脳型を呈する変性疾患としての皮質性小脳萎縮症は、実際には非常に少ないと考えられる。 |
|
| |
|
| {| class="wikitable"
| | ===症候=== |
| |+表4. 潜性 (劣性) 遺伝の脊髄小脳変性症
| | 中年期以降に、小脳性の[[体幹運動失調]]と[[構音障害]]が緩徐に進行する。経過は多系統萎縮症に比べて緩やかであり、進行しても独立歩行が可能な例もある。四肢の協調運動障害も次第に進行するが、小脳系以外の症候は認めない。 |
| !疾患!!原因遺伝子/遺伝子座!!Phenotype MIM number!!遺伝子変異および臨床的特徴
| |
| |-
| |
| | [[Friedreich失調症]]([[FRDA]]) || [https://www.omim.org/entry/606829 FXN ]|| [https://www.omim.org/entry/229300 229300] || 日本では報告なし、イントロンGAAリピート伸長、腱反射低下、[[深部感覚]]障害、[[心筋症]]
| |
| |-
| |
| | [[ataxia-telangiectasia]] (AT) || [https://www.omim.org/entry/607585 ATM ] || [https://www.omim.org/entry/208900 208900 ]|| 毛細血管拡張、[[免疫不全]]、[[悪性腫瘍]]合併
| |
| |-
| |
| | [[ATLD1]] || [https://www.omim.org/entry/600814 MRE11A ] || [https://www.omim.org/entry/604391 604391] || 眼球運動失行、ATに類似だが毛細血管拡張、免疫不全はない
| |
| |-
| |
| | [[ATLD2]] || [https://www.omim.org/entry/176740 PCNA ] || [https://www.omim.org/entry/615919 615919] || 発達障害、感音性難聴、低身長、毛細血管拡張
| |
| |-
| |
| | [[EAOH]]/[[AOA1]] || [https://www.omim.org/entry/606350 APTX ] || [https://www.omim.org/entry/208920 208920] || 日本でFRDAと報告されていた症例の多く、[[眼球運動]]失行、[[低アルブミン血症]]、[[高コレステロール血症]]、末梢神経障害、認知機能障害
| |
| |-
| |
| | [[SCAN2]]/[[AOA2]]/[[SCAR1]] || [https://www.omim.org/entry/608465 SETX ] || [https://www.omim.org/entry/606002 606002 ]|| [[α-フェトプロテイン]]上昇、眼球運動失行、末梢神経障害
| |
| |-
| |
| | [[AOA3]] || [https://www.omim.org/entry/611317 PIK3R5 ] || [https://www.omim.org/entry/615217 615217 ]|| サウジアラビアの家系、20歳代発症、眼球運動失行、末梢神経障害
| |
| |-
| |
| | [[AOA4]] || [https://www.omim.org/entry/605610 PNKP ] || [https://www.omim.org/entry/616267 616267 ]|| [[シャルコー・マリー・トゥース病]], [[シャルコー・マリー・トゥース病#病態生理|type 2B2]]と同じ遺伝子、ジストニア、眼球運動失行、末梢神経障害
| |
| |-
| |
| | [[SCAR2]] || [https://www.omim.org/entry/613036 PMPCA ] || [https://www.omim.org/entry/213200 213200 ]|| 精神発達障害、運動発達障害、[[白内障]]、企図振戦
| |
| |-
| |
| | [[SCAR3]] || 6p23-p21 || [https://www.omim.org/entry/271250 271250 ]|| [[視神経]]・[[蝸牛]]変性、[[レフサム病]]に類似
| |
| |-
| |
| | [[SCAR4]] || [https://www.omim.org/entry/608877 VPS13D] || [https://www.omim.org/entry/607317 607317 ]|| 痙性歩行、腱反射亢進、発症年齢は小児期~成人、軽度認知機能障害
| |
| |-
| |
| | [[GAMOS1]]/[[SCAR5]] || [https://www.omim.org/entry/616144 WDR73 ] || [https://www.omim.org/entry/251300 251300 ]|| 発達障害、精神発達障害、[[痙性失調]]、[[小頭症]]、視神経萎縮
| |
| |-
| |
| | [[SCAR6]] || 20q11-q13 || [https://www.omim.org/entry/608029 608029 ]|| ノルウェーの家系、乳児発症、非進行性、筋トーヌス低下、知能は正常
| |
| |-
| |
| | [[SCAR7]] || [https://www.omim.org/entry/607998 TPP1 ] || [https://www.omim.org/entry/609270 609270 ]|| [[セロイドリポフスチン症2型]]と同一遺伝子、症状は軽症~重度まで差があり、腱反射亢進、後索障害による深部感覚障害
| |
| |-
| |
| | [[SCAR8]] || [https://www.omim.org/entry/608441 SYNE1 ] || [https://www.omim.org/entry/610743 610743 ]|| 発症は20~30歳代、痙性や二次性の筋骨格系変形を伴うこともある
| |
| |-
| |
| | [[COQ10D4]]/[[SCAR9]] || [https://www.omim.org/entry/606980 ADCK3 ] || [https://www.omim.org/entry/612016 612016 ]|| 運動不耐性、痙攣や軽度知的障害を合併することがある、経口[[補酵素Q10]]は無効
| |
| |-
| |
| | [[SCAR10]] || [https://www.omim.org/entry/613726 ANO10 ] || [https://www.omim.org/entry/613728 613728 ]|| 筋内補酵素Q10が低下する症例あり、補酵素Q10が部分的に有効
| |
| |-
| |
| | [[SCAR11]] || [https://www.omim.org/entry/610949 SYT14 ] || [https://www.omim.org/entry/614229 614229 ]|| 日本から1家系(兄弟例)、精神運動障害
| |
| |-
| |
| | [[SCAR12]] || [https://www.omim.org/entry/605131 WWOX ] || [https://www.omim.org/entry/614322 614322 ]|| developmental and epileptic encephalopathy-28と同じ遺伝子、全身けいれん、精神運動障害
| |
| |-
| |
| | [[SCAR13]] || [https://www.omim.org/entry/604473 GRM1 ] || [https://www.omim.org/entry/614831 614831 ]|| 乳児期の精神運動障害、知的障害、脳室拡大
| |
| |-
| |
| | [[SCAR14]] || [https://www.omim.org/entry/604985 SPTBN2 ] || [https://www.omim.org/entry/615386 615386 ]|| 精神運動発達障害
| |
| |-
| |
| | [[SCAR15]] || [https://www.omim.org/entry/613516 RUBCN ] || [https://www.omim.org/entry/615705 615705 ]|| 知的障害、発達障害、てんかん
| |
| |-
| |
| | [[SCAR16]] || [https://www.omim.org/entry/607207 STUB1 ] || [https://www.omim.org/entry/615768 615768 ]|| 下肢の痙性、感覚性末梢神経障害
| |
| |-
| |
| | [[SCAR17]] || [https://www.omim.org/entry/616120 CWF19L1 ] || [https://www.omim.org/entry/616127 616127 ]|| 知的障害
| |
| |-
| |
| | [[SCAR18]] || [https://www.omim.org/entry/602368 GRID2 ] || [https://www.omim.org/entry/616204 616204 ]|| 精神運動発達障害、腱反射亢進
| |
| |-
| |
| | [[LIKNS]]/[[SCAR19]] || [https://www.omim.org/entry/107310 SLC9A1 ] || [https://www.omim.org/entry/616291 616291 ]|| 重度の感音性難聴
| |
| |-
| |
| | [[SCAR20]] || [https://www.omim.org/entry/616105 SNX14 ] || [https://www.omim.org/entry/616354 616354 ]|| 重度の精神運動発達障害、小頭症、鼻翼が広い、厚い口唇、感音性難聴、けいれん
| |
| |-
| |
| | [[SCAR21]] || [https://www.omim.org/entry/607982 SCYL1 ] || [https://www.omim.org/entry/616719 616719 ]|| 肝障害、末梢神経障害、軽度知的障害
| |
| |-
| |
| | [[SCAR22]] || [https://www.omim.org/entry/614884 VWA3B ] || [https://www.omim.org/entry/616948 616948 ]|| 日本の1家系、知的障害、腱反射亢進、痙性
| |
| |-
| |
| | [[SCAR23]] || [https://www.omim.org/entry/605764 TDP2 ] || [https://www.omim.org/entry/616949 616949 ]|| てんかん、知的障害
| |
| |-
| |
| | [[SCAR24]] || [https://www.omim.org/entry/610552 UBA5 ] || [https://www.omim.org/entry/617133 617133 ]|| 中国の1家系、成長が遅い、白内障、知能は正常
| |
| |-
| |
| | [[SCAR25]] || [https://www.omim.org/entry/604261 ATG5 ] || [https://www.omim.org/entry/617584 617584 ]|| トルコの1家系、精神運動発達障害
| |
| |-
| |
| | [[SCAR26]] || [https://www.omim.org/entry/194360 XRCC1 ] || [https://www.omim.org/entry/617633 617633 ]|| 祖先が東インドの患者1名、28歳歩行障害で発症、眼球運動失行、末梢神経障害
| |
| |-
| |
| | [[SCAR27]] || [https://www.omim.org/entry/618128 GDAP2 ] || [https://www.omim.org/entry/618369 618369 ]|| 成人発症、認知機能障害
| |
| |-
| |
| | [[SCAR28]] || [https://www.omim.org/entry/618802 THG1L ] || [https://www.omim.org/entry/618800 618800 ]|| 痙性、軽度知的障害
| |
| |-
| |
| | [[SACS]]/[[ARSACS]] || [https://www.omim.org/entry/604490 SACS ] || [https://www.omim.org/entry/270550 270550 ]|| 末梢神経障害、網膜色素線条、知的障害を認めることもある、腱反射は低下~亢進、末梢神経障害
| |
| |-
| |
| | [[SCAN1]] || [https://www.omim.org/entry/607198 TDP1 ] || [https://www.omim.org/entry/607250 607250 ]|| サウジアラビアの1家系、末梢神経障害、低アルブミン血症、高コレステロール血症
| |
| |}
| |
|
| |
|
| ATLD= [[ataxia-telangiectasia-like disorder]], EAOH=[[early onset ataxia with ocular motor apraxia and hypoalbuminemia]], AOA=[[ataxia- ocular motor apraxi]]a, SCAN= [[spinocerebellar ataxia with axonal neuropathy]], SCAR=[[spinocerebellar ataxia, autosomal recessive]], GAMOS1= [[Galloway-Mowat syndrome 1]], COQ10D4=[[coenzyme Q10 deficiency, primary, 4]], LIKNS=[[Lichtenstein-Knorr syndrome]], SACS=[[Spastic ataxia, Charlevoix-Saguenay type]], ARSACS=[[autosomal recessive spastic ataxia of Charlevoix-Saguenay]]
| | ===補助診断法=== |
| | [[image:脊髄小脳変性症2.png|thumb|350px|'''図2.皮質性小脳萎縮症のMRIにおける小脳萎縮'''<br>小脳の萎縮を認めるが、脳幹は保たれている]] |
|
| |
|
| [[ファイル:Yokoseki spinocerebellar degeneration Fig2.png|サムネイル|'''図2. 脊髄小脳変性症と変性部位'''<br>赤印が、障害部位を示し、濃い印は障害の程度が強いことを示す。参考文献<ref name=Tada2015 />より改変。]] | | 画像検査では、小脳に限局して進行性の萎縮を認める(図2)。病初期には[[虫部]]前葉から萎縮が始まり、次第に小脳半球に波及する。しかし、[[wikipedia:ja:甲状腺機能低下症|甲状腺機能低下症]]、[[wikipedia:ja:ビタミンE欠乏症|ビタミンE欠乏症]]、[[wikipedia:ja:ビタミンB1欠乏症|ビタミンB1欠乏症]]、[[wikipedia:ja:Wilson病|Wilson病]]などの代謝性疾患、[[慢性アルコール中毒]]、[[フェニトイン]]や[[臭化バレリル尿素]]などの薬物中毒、[[有機水銀中毒]]、[[wikipedia:ja:トルエン|トルエン]]や[[wikipedia:ja:ベンゼン|ベンゼン]]などの[[wikipedia:ja:有機溶媒中毒|有機溶媒中毒]]、[[傍腫瘍性小脳変性症]]([[腫瘍随伴性神経症候群]])、[[グルテン失調症]]、[[GAD抗体陽性失調症]]、[[急性小脳炎]]、[[Fisher症候群]]、[[神経Behçet病]]、[[多発性硬化症]]、小脳血管障害、小脳腫瘍など、多くの疾患を除外する必要があり、診断を皮質性小脳萎縮症と確定することは容易ではない。 |
| [[ファイル:Yokoseki spinocerebellar degeneration Fig3.png|サムネイル|'''図3. 大脳小脳の神経連絡'''<br>破線は小脳への入力、実線は小脳からの出力を示す。]] | |
| [[ファイル:Yokoseki spinocerebellar degeneration Fig4.png|サムネイル|'''図4. 脊髄小脳の神経連絡'''<br>破線は小脳への入力、実線は小脳からの出力を示す。]] | |
| [[ファイル:Yokoseki spinocerebellar degeneration Fig5.png|サムネイル|'''図5. 前庭小脳の神経連絡'''<br>破線は小脳への入力、実線は小脳からの出力を示す。]] | |
|
| |
|
| == 症状と病態生理 == | | ===治療=== |
| 人が動作をする際には、小脳による運動制御が必須であるが、その制御を行う際には、脳内に[[内部モデル]]を必要とする「[[内部モデル仮説]]」が提唱されている<ref name=Wolpert1998><pubmed>21227230</pubmed></ref> 。運動に必要な内部モデルの形成、情報処理、最適化において、小脳の役割が重要であると考えられている。
| | 根治的な治療法は確立されていないが、小脳の機能維持を目的として、四肢末梢への錘負荷やバランス訓練などのリハビリテーションが広く行われてきた。小脳が正常に保たれている脳血管障害に対する機能回復訓練とは異なり、[[運動学習]]の首座と考えられる小脳に進行性の変性が起きている小脳変性症の場合にも、繰り返し学習による可塑性(use- dependent plasticity)が獲得されるか否かは明らかでなかった。そこで、厚生労働省の運動失調症調査研究班で筆者らは、短期集中リハビリが小脳性運動失調の進行抑制に有効であるかを検証する臨床治験を、皮質性小脳萎縮症と遺伝性純粋小脳型失調症(SCA6とSCA31)を対象として実施し、1日各1時間の理学療法と作業療法を1ヶ月間継続すると、小脳性運動失調は改善し、その効果は最大6ヶ月続くことが実証された。この効果は既存の薬物治療効果を上回っており、小脳機能維持を目的としたリハビリテーション体制を整備することが今後の課題である。 |
|
| |
|
| 脊髄小脳変性症では、小脳や小脳につながる神経路の障害により運動失調を来すが、疾患により変性部位に差違がある ('''表5'''、'''図2''') <ref name=Tada2015><pubmed>25637456</pubmed></ref> 。つまり、障害される解剖学的部位により、小脳失調を生じる機序が異なると考えられる。一方現在の診察方法では、小脳症状は後述の通り表面的な症状でしか評価ができないため、'''表5'''に示すような障害されている部位を推定することは困難である。今後小脳症状の機能解析の新たな開発により、障害部位特異的な小脳症状の評価が可能となり、さらにはリハビリテーションを始めとする疾患特異的な治療法の開発が期待される。
| | ==遺伝性脊髄小脳変性症== |
| | ===常染色体優性遺伝性脊髄小脳変性症=== |
|
| |
|
| {| class="wikitable" style="text-align:center;"
| | ====概念==== |
| |+表5. 各脊髄小脳変性症の変性部位 (参考文献<ref name=Tada2015/>より改変)
| | 遺伝性脊髄小脳変性症の9割以上を占める[[常染色体優性遺伝性脊髄小脳変性症]](Autosomal dominant SCD:ADSCD)は、その約9割まで原因遺伝子が同定された。原因遺伝子座が同定された常染色体優性遺伝性脊髄小脳変性症は、脊髄小脳失調症(spinocerebellar ataxia:SCA)の何番というように、病名を機械的に決める方式が広く採用されている。The Human Genome Organization(HUGO)には現在[[SCA41]]まで登録されており、このうちSCA9、16、22は欠番である。一方、わが国で頻度が高い[[歯状核赤核淡蒼球ルイ体萎縮症]]([[dentatorubral pallidoluysian atrophy]]:[[DRPLA]])は、脊髄小脳失調症としては登録されていない。 |
| !障害される機能!!colspan="3"|感覚系からのフィードバック情報の障害!!大脳からの入力障害!! !!内部モデル本体の障害!!出力の障害!!誤差情報の障害
| |
| |-
| |
| ! 変性部位 !! [[後根神経節]] !! [[クラーク柱]] !! [[前庭神経核]] !! [[橋核]] !! [[顆粒細胞]] !! [[プルキンエ細胞]] !! [[歯状核]] !! [[下オリーブ核]]
| |
| |-
| |
| | DRPLA|| - || - || - || - || - || + || +++ || -
| |
| |-
| |
| | SCA6 || - || - || - || - || + || +++ || ++ || +
| |
| |-
| |
| | SCA31 || - || - || - || - || - || +++ || - || -
| |
| |-
| |
| | MSA || - || - || + || +++ || - || + || - || +++
| |
| |-
| |
| | SCA2 || - || - || ++ || +++ || +++ || +++ || ++ || +++
| |
| |-
| |
| | SCA1 || - || ++ || + || + || ++ || ++ || ++ || ++
| |
| |-
| |
| | MJD || - || +++ || ++ || ++ || - || + || +++ || -
| |
| |-
| |
| | EAOH || +++ || +++ || - || - || + || +++ || + || +
| |
| |}
| |
| DRPLA=dentatorubral-pallidoluysian atrophy、SCA6=spinocerebellar ataxia 6、SCA31=spinocerebellar ataxia 31、MSA=multiple system atrophy、SCA2=spinocerebellar ataxia 2、SCA1=spinocerebellar ataxia 1、MJD=Machado-Joseph disease、EAOH=early onset ataxia with ocular motor apraxia and hypoalbuminemia<br>
| |
| 変性の程度を重度(+++)、中等度(++)、軽度(+)で示す。
| |
|
| |
|
| 小脳やその経路に起因する症状、個々の疾患に合併する他の神経部位に起因する症状 (末梢神経障害や自律神経障害など) を認める。小脳失調は、障害される部位により、様々な症状である<ref name=筧慎治2015>'''筧慎治, 石川享宏, 本多武尊, 三苫博 (2015)'''<br> 【小脳の最新知見-基礎研究と臨床の最前線】小脳は何をしているか 構造と機能の最先端 小脳の機能 平衡、協調運動機能. 医学のあゆみ. 255, 947-954</ref> 。
| | わが国では[[Machado-Joseph病]]([[MJD]]:別名[[SCA3]])の頻度が最も高く、全体の約4分の1を占める。[[SCA6]]、DRPLA、[[SCA31]]がこれに次ぐ。これらの頻度には地域差があり、東日本ではMachado-Joseph病、西日本ではSCA6が多い。 |
| === 大脳小脳 (小脳半球外側部) の障害 ===
| |
| [[小脳半球]]外側部と[[歯状核]]からなる[[大脳小脳]]は、[[脊髄]]などの末梢から直接入力は少なく、大脳皮質から[[橋]]を経由して入力を受ける。出力は、[[歯状核]]を起始部として、[[視床]]や[[赤核]]へ入力される。視床から大脳皮質広範囲に投射することにより、四肢の遠位筋の運動制御が行われる。そのため、同部位の障害では、[[運動分解]] (decomposition) や、[[測定異常]] (dysmetria) 、[[反復拮抗運動不能]] (adiadochokinesis) など四肢の運動症状を認める ('''図3''')。
| |
| === 脊髄小脳(小脳虫部、小脳半球内側部)の障害 ===
| |
| [[小脳虫部]]および小脳半球内側部と[[中位核]] ([[球状核]]、[[栓状核]]) からなる[[脊髄小脳]]は、脊髄からの[[触覚]]、[[圧覚]]、[[位置覚]]などの[[体性感覚]]の情報入力を受け、[[前庭]]や[[網様体]]へ投射し、[[前庭脊髄路]]、[[網様体脊髄路]]を経由して脊髄に出力される。これらの出力は、最終的に[[体幹筋]]に支配し[[姿勢制御]]に重要であり、脊髄小脳の障害では、小脳性の体幹失調や歩行障害を起こす ('''図4''')。
| |
| === 前庭小脳 (片葉小節葉) の障害 ===
| |
| [[前庭小脳]] ([[片葉小節葉]]) は、[[三半規管]]や[[耳石器]]などの前庭から入力を受ける。また、[[中脳]]および[[視覚野]]から視覚入力も受ける。前庭小脳からの出力は、[[前庭神経核]]に投射される。同部は、前庭眼反射の制御を行っており、障害されることにより[[眼振]]、眼球測定異常、前庭眼反射の異常などの眼球運動障害を起こす ('''図5''')。
| |
|
| |
|
| == 治療 ==
| | 常染色体優性遺伝性脊髄小脳変性症における遺伝子異常の多くは、翻訳領域に存在するCAGリピート長が正常の2、3倍に異常伸長していることであり、遺伝子レベルでは[[CAGリピート病]]、タンパク質レベルでは[[ポリグルタミン病]]とよばれる。伸長したポリグルタミン鎖を含むタンパク質が凝集する過程で形成されるオリゴマーに細胞障害性があると考えられる。 |
| 脊髄小脳変性症の根本的治療法は、いまだ確立されていない。
| |
|
| |
|
| 本邦で小脳失調症状の改善を目的として、[[甲状腺刺激ホルモン放出ホルモン]] ([[thyrotropin releasing hormone]], [[TRH]]) 誘導体が使用されている。TRH誘導体使用の経緯は、1970年代初頭に開発された失調症モデルマウスである[[rolling mouse Nagoya]]の解析に由来する<ref name=織田鉄一1973>'''織田鉄一 (1973)'''<br>歩行異常マウスの発見と維持. 実験動物. 22, 281-288</ref> 。このマウスは、のちに[[電位依存性カルシウムチャネル#Cav2_.28N.2C_P.2FQ.2C_R.E5.9E.8B.29|P/Q-type Ca<sup>2+</sup> channels]]に変異を有していることが明かとなっている<ref name=Mori2000><pubmed>10908603</pubmed></ref> 。このrolling mouse Nagoyaでは、小脳脳幹部のTRH含有量が減少していること<ref name=祖父江逸郎1977>'''祖父江逸郎 (1977)'''<br> 視床下部ホルモンと中枢神経機能との新しい接点TRHと運動失調との関連をめぐって. 臨床神経学. 17, 791-799</ref> 、小脳ではノルアドレナリン神経終末に異常があることが報告され<ref name=Adachi1975>'''Adachi, K. (1975).'''<br>Changes in the cerebellar noradrenaline nerve terminals of the neurological murine mutant rolling mouse Nagoya : A histofluorescence analysis. Neurobiol and Neurophys. 3, 329-330 [https://ci.nii.ac.jp/naid/10030205506/ CiNii]</ref> 、TRHは[[ノルアドレナリン]]のturnoverに関与することなどから、TRH投与が失調症状改善につながると可能性が期待された。実際にrolling mouse Nagoya にTRHを投与により失調症状に有効であることが示され<ref name=小長谷正明1980>'''小長谷正明, 高柳哲也, 室賀辰夫他 (1980)'''<br>RollingmouseNagoyaの脳内ノルアドレナリン代謝とthyrotropinreleasinghormoneの影響. 臨床神経学. 20, 181-188</ref> 、その後本邦の多施設での治験を経て1985年に点滴用TRH製剤である[[プロチレリン酒石酸]]が脊髄小脳変性症の運動失調症状の改善として保険適応となり<ref name=祖父江1982>'''祖父江逸郎, 高柳哲也, 中西孝 (1982).'''<br> 脊髄小脳変性症に対するThyrotropin Releasing Hormone Tartrateの治療研究 二重盲検比較対照臨床試験による検討. 神経研究の進歩. 26, 1190-1214</ref> 、2000年以降は内服のTRH製剤である[[タルチレリン水和物]]が脊髄小脳変性症の運動失調の改善を目的として使用されている<ref name=金澤一郎1997>'''金澤一郎, 里吉栄二郎, 平山惠造他 (1997).'''<br>Taltirelin hydrate(TA-0910)の脊髄小脳変性症に対する臨床評価 プラセボを対照とした臨床第III相二重盲検比較試験. 臨床医薬. 13, 4169-4224</ref> 。タルチレリン水和物の失調症状の改善効果は極めて限定的であり、現在より効果の強いTRH製剤の開発が行われている<ref name=Nishizawa2020><pubmed>31937586</pubmed></ref> 。
| | ポリグルタミン病では、世代を経る毎に発症年齢が若年化し、重症化する表現促進現象(anticipation)が認められる。Mendel遺伝では説明できない現象であったが、リピート数の伸長によることが明らかになっている。翻訳領域のCAGリピートは父方から伝搬する場合に著明に伸長する傾向があり、CAGリピート数が短いSCA6を除き、発症年齢とリピート数には負の相関が認められる。 |
|
| |
|
| 脊髄小脳変性症などの神経変性疾患では、運動機能の維持、合併症の予防目的にリハビリテーションが実施されている。脊髄小脳変性症において、短期集中リハビリテーションが有効であることを日本の「運動失調症の病態解明と治療法開発に関する研究」を中心とする研究班での研究により示されている。このリハビリテーションの研究 (Trial for Cerebellar Ataxia Rehabilitation, CAR trial) では、小脳症状が主体であるSCA6、SCA31、皮質性小脳萎縮症患者に対して、1日1~2時間、週3~7回、4週間のバランスや歩行を中心とした短期集中リハビリテーションにより、運動失調や歩行の改善を認め、かつその効果が半年~1年継続することが示されている<ref name=Miyai2012><pubmed>22140200</pubmed></ref> 。
| | 遺伝性脊髄小脳変性症に関する遺伝子診断を行う際には、[[wikipedia:ja:文部科学省|文部科学省]]、厚生労働省、[[wikipedia:ja:経済産業省|経済産業省]]の3省庁合同のヒトゲノム・遺伝子解析研究に関する最新の倫理指針を遵守する必要がある。根治的な治療法が確立されていない遺伝性疾患の[[発症前診断]]や[[保因者診断]]は、原則として行わない。 |
|
| |
|
| また現在まで、非常に種々の薬物等で治験が実施されており、現在も治験が進行中である ('''表6''') 。一部は部分的に効果を認めているが、多くは効果を示すことができていない。今後は、遺伝性脊髄小脳変性症に対する核酸医療の開発も期待されている。
| | ====各論==== |
| | わが国で頻度の高い病型を中心とし、その他の病型は表2に一括した。 |
|
| |
|
| {| class="wikitable"
| | #Machado-Joseph病 MJD(SCA3)<br> Machado-Joseph病は当初、[[wikipedia:ja:ポルトガル|ポルトガル]]領[[wikipedia:ja:アゾレス諸島|アゾレス諸島]]から北米に移民した子孫の間に見出された疾患であり、その後、欧州で記載されたSCA3でも同一のCAGリピート伸長が確認されている。臨床的にはRosenbergにより、若年発症で[[錐体路症状]]と、[[ジストニア]]などの[[錐体外路症状]]が目立つ1型、成年発症で[[痙性失調症]]と[[眼振]]を呈する2型、高齢発症で[[筋萎縮]]や[[末梢神経障害]]などの末梢性病変を伴う3型、[[パーキンソニズム]]を伴うまれな4型に分けられている。[[Ataxin3]]遺伝子に存在するCAGリピートの伸長は1型で最も長く、3型では短い。顔面筋の線維束性収縮や[[ミオキミア]]、[[びっくり眼]]などはMachado-Joseph病によくみられる。 |
| |+表6. 脊髄小脳変性症の治験
| | #SCA6<br> 50歳前後で発症し、小脳性運動失調症状のみを呈する純粋小脳型常染色体優性遺伝性脊髄小脳変性症であり、[[P/Q型電位依存性Caチャネル]]α1サブユニット遺伝子のC末端に位置するCAGリピートの軽度の伸長による。同遺伝子の点変異は、[[反復発作性運動失調症2型]](episodic ataxia type 2: EA2)と[[家族性片麻痺性片頭痛]]の原因でもある。 |
| !化合物!!ClinicalTrials.gov Identifier !! 年 !! 国 !! 対象疾患 !! Phase !! 結果
| | #SCA31<br> 常染色体優性遺伝性脊髄小脳変性症では最も高齢の60歳前後で発症する純粋小脳型常染色体優性遺伝性脊髄小脳変性症であるが、遺伝子診断によらずにSCA6と鑑別することは困難である。わが国では長野県、静岡県、鹿児島県で特に多い。第16染色体長腕の[[BEAN]]と[[TK2]]遺伝子に共通するイントロンに挿入されたTGGAAという5塩基リピートが著明に伸長しており、転写産物によるRNA fociが形成されていることから、これと相互作用する核タンパク質の機能変化が想定される。 |
| |-
| | #DRPLA<br> わが国に多い常染色体優性遺伝性脊髄小脳変性症で、発症年齢により臨床症状が異なる。[[atrophin 1]]遺伝子に存在するCAGリピートが長い場合は若年発症となり、[[進行性ミオクローヌスてんかん]]の臨床像を示す。伸長の程度が軽い場合には成人発症となり、[[認知]]機能障害や[[不随意運動]]などを呈する。ポリグルタミン病では最も著明な[[表現促進現象]]がみられ、リピート伸長の程度により、発症年齢や臨床像、重症度が規定される。[[小脳歯状核]]とその遠心路、[[淡蒼球]][[視床下核]]系に変性と萎縮を認めるだけでなく、[[大脳白質]]にも広範な変性像が認められる。 |
| | [[Taltirelin Hydrate]] || [https://www.clinicaltrials.gov/ct2/show/NCT04107740 NCT04107740] || 2019- || 韓国 || SCD全般 || 4 || 現在進行中
| | #毛細血管拡張運動失調症(ataxia telangiectasia:AT;Louis-Bar症候群)<br> 幼児期に小脳性運動失調と皮膚や眼球結膜の[[wikipedia:ja:毛細血管|毛細血管]]拡張症で発症する。[[wikipedia:ja:IgA|IgA]]が低下し、[[wikipedia:ja:免疫不全|免疫不全]]のために感染症を起こしやすく、また高率に[[wikipedia:ja:悪性リンパ腫|悪性リンパ腫]]などの悪性腫瘍を合併する。ATの責任遺伝子[[ATM]]は2本鎖DNAの損傷修復に関与するタンパク質をコードする。神経症状として眼球運動失行を認め、以下に述べる[[aprataxin]]や[[senataxin]]の欠損症と病態、臨床症候は類似している。 |
| |-
| |
| | [[KPS-0373]] ([[Rovatirelin]]) || [https://www.clinicaltrials.gov/ct2/show/NCT01970098 NCT01970098] || 2013-2015 || 日本 || 軽度~中等度のSCD || 3 || [https://jnnp.bmj.com/content/91/3/254 部分的に有効]
| |
| |-
| |
| | [[Riluzole]] || [https://www.clinicaltrials.gov/ct2/show/NCT03347344?cond=Spinocerebellar+Degeneration&draw=3&rank=11 NCT03347344] || 2018-2019 || フランス || SCA2 || 3 || 不明
| |
| |-
| |
| | [[Transcranial Magnetic Stimulation]] || [https://www.clinicaltrials.gov/ct2/show/NCT03347344 NCT01975909] || 2013-2016 || 米国 || genetically-confirmed SCA || Not Applicable || 有効性なし
| |
| |-
| |
| | [[Lithium Carbonate]] || [https://www.clinicaltrials.gov/ct2/show/NCT00683943 NCT00683943] || 2008-2010 || 米国 || SCA1 || 1 ||
| |
| |-
| |
| | [[Intravenous Immune Globulin]] || [https://www.clinicaltrials.gov/ct2/show/NCT02287064 NCT02287064] || 2015-2016 || 米国 || SCA types 1, 2, 3, 6, 10, or 11 || 1 || まだ発表なし
| |
| |-
| |
| | Oral [[Trehalose]] || [https://www.clinicaltrials.gov/ct2/show/NCT04399265 NCT04399265] || 2020ー- || マレーシア || Genetically confirmed SCA 3 || Not Applicable || 進行中
| |
| |-
| |
| | [[Varenicline]] ([[Chantix®]]) || [https://www.clinicaltrials.gov/ct2/show/NCT00992771 NCT00992771] || 2009-2012 || 米国 || SCA3 || 2 || [https://n.neurology.org/content/78/8/545.long 有効] | |
| |-
| |
| | [[Umbilical Cord Mesenchymal Stem Cells]] || [https://www.clinicaltrials.gov/ct2/show/NCT03378414 NCT03378414] || 2017- || 中国 || SCA1,2,4,6 || 2 || 詳細不明
| |
| |-
| |
| | [[Troriluzole]] || [https://www.clinicaltrials.gov/ct2/show/NCT02960893 NCT03701399] || 2018- || 米国 || SCA1,2,3,6,7,8,10 || 2,3 || 進行中
| |
| |-
| |
| | [[Sodium Phenylbutyrate]] || [https://www.clinicaltrials.gov/ct2/show/NCT01096095 NCT01096095] || 2010-2012 || ブラジル || SCA3 || 2 || 中止
| |
| |-
| |
| | [[Dalfampridine]] || [https://www.clinicaltrials.gov/ct2/show/NCT01811706 NCT01811706] || 2013-2015 || 米国 || SCA1,2,3,6 || Not Applicable || 有効性なし
| |
| |-
| |
| | [[CAD-1883]] || [https://www.clinicaltrials.gov/ct2/show/NCT04301284 NCT04301284] || 2020- || 米国 || SCA1,2,3,6,7,10,17 || 2 || 延期(COVID-19のため
| |
| |-
| |
| | [[Docosahexaenoic Acid]] ([[DHA]]) || [https://www.clinicaltrials.gov/ct2/show/NCT03109626 NCT03109626] || 2015-2018 || イタリア || SCA38 || Not Applicable || [https://onlinelibrary.wiley.com/doi/full/10.1002/ana.25059 有効]
| |
| |-
| |
| | [[Stemchymal®]] || [https://www.clinicaltrials.gov/ct2/show/NCT02540655 NCT02540655] || 2015- || 台湾 || SCA2,3 || 2 || 不明
| |
| |-
| |
| | Intravenous [[Cabaletta]] || [https://www.clinicaltrials.gov/ct2/show/NCT02147886 NCT02147886] || 2014-2016 || イスラエル || SCA3 || 2 || 結果?
| |
| |-
| |
| | [[Coenzyme Q10]] || [https://www.clinicaltrials.gov/ct2/show/NCT00957216 NCT00957216] || 2009-2018 || 米国 || 弧発性SCD || 1 || 結果?
| |
| |-
| |
| | [[BHV-4157]] || [https://clinicaltrials.gov/ct2/show/NCT03408080 NCT03408080] || 2018- || 米国 || SCA1,2,3,6 || 3 || エントリー中止中
| |
| |-
| |
| | [[Vatiquinone]] || [https://www.clinicaltrials.gov/ct2/show/NCT04577352 NCT04577352] || 2020- || 米国 || Friedreich Ataxia || 2,3 || 開始前
| |
| |-
| |
| | [[CTI-1601]] || [https://www.clinicaltrials.gov/ct2/show/NCT04519567 NCT04519567] || 2020- || 米国 || Friedreich Ataxia || 1 || 開始前
| |
| |-
| |
| | [[Etravirine]] || [https://clinicaltrials.gov/ct2/show/NCT04273165 NCT04273165] || 2020- || イタリア || Friedreich Ataxia || 2 || 開始前
| |
| |-
| |
| | [[RT001]] || [https://www.clinicaltrials.gov/ct2/show/NCT04102501 NCT04102501] || 2019- || 米国 || Friedreich Ataxia || 3 || 進行中
| |
| |-
| |
| | [[Resveratrol]] || [https://clinicaltrials.gov/ct2/show/NCT03933163 NCT03933163] || 2019- || 米国 || Friedreich Ataxia || 2 || 進行中
| |
| |-
| |
| | [[MIN-102]] || [https://clinicaltrials.gov/ct2/show/NCT03917225 NCT03917225] || 2019- || EU || Friedreich Ataxia || 2 || リクルート終了
| |
| |-
| |
| | [[γ-interferon]] || [https://clinicaltrials.gov/ct2/show/NCT03888664 NCT03888664]<br>[https://clinicaltrials.gov/ct2/show/NCT02797080 NCT02797080]<br>[https://clinicaltrials.gov/ct2/show/NCT02593773 NCT02593773]<br>[https://www.clinicaltrials.gov/ct2/show/NCT01965327 NCT01965327]|| || || Friedreich Ataxia || 2、3 ||
| |
| |-
| |
| | [[Nicotinamide]] || [https://clinicaltrials.gov/ct2/show/NCT03761511 NCT03761511] || 2018- || EU || Friedreich Ataxia || 2 || 進行中
| |
| |-
| |
| | [[TAK-831]] || [https://www.clinicaltrials.gov/ct2/show/NCT03214588 NCT03214588] || 2017-2019 || 米国 || Friedreich Ataxia || 2 || 有効性なし
| |
| |-
| |
| | [[Rosuvastatin]] || [https://www.clinicaltrials.gov/ct2/show/NCT02705547 NCT02705547] || 2016-2018 || 米国 || Friedreich Ataxia || 1 || リクルート終了
| |
| |-
| |
| | [[(+)-Epicatechin]] || [https://clinicaltrials.gov/ct2/show/NCT02660112 NCT02660112] || 2016-2019 || 米国 || Friedreich Ataxia || 2 || 有効性なし
| |
| |-
| |
| | [[Methylprednisolone]] || [https://www.clinicaltrials.gov/ct2/show/NCT02424435 NCT02424435] || 2015-2020 || 米国 || Friedreich Ataxia || 1 || リクルート終了
| |
| |-
| |
| | [[EPI-743]] ([[α-tocotrienol quinone]]) || [https://clinicaltrials.gov/ct2/show/NCT01962363 NCT01962363] || 2013-2016 || 米国 || Friedreich's Ataxia with Point Mutations || 2 || [https://n.neurology.org/content/86/16_Supplement/P5.388 効果あり]
| |
| |-
| |
| | [[Omaveloxolone]] || [https://clinicaltrials.gov/ct2/show/NCT02255435 NCT02255435] || 2014- || 米国 || Friedreich Ataxia || 2 || リクルート停止
| |
| |-
| |
| | [[Acetyl-L-Carnitine]] || [https://www.clinicaltrials.gov/ct2/show/NCT01921868 NCT01921868] || 2013-2017 || 米国 || Friedreich Ataxia || Not Applicable || 結果未報告
| |
| |-
| |
| | [[VP 20629]] || [https://clinicaltrials.gov/ct2/show/NCT01898884 NCT01898884] || 2013-2016 || 米国 || Friedreich Ataxia || 1 || リクルート終了
| |
| |-
| |
| | [[EPI-743]] || [https://clinicaltrials.gov/ct2/show/NCT01728064 NCT01728064] || 2012-2015 || 米国 || Friedreich Ataxia || 2 || 有効性なし
| |
| |-
| |
| | [[Bupropion]] and [[Citalopram]] || [https://clinicaltrials.gov/ct2/show/NCT01716221 NCT01716221] || 2012-2016 || 米国 || Friedreich Ataxia || 4 || 終了
| |
| |-
| |
| | [[Resveratrol]] || [https://clinicaltrials.gov/ct2/show/NCT01339884 NCT01339884] || 2011-2014 || オーストラリア || Friedreich Ataxia || 1,2 || 終了
| |
| |-
| |
| | [[Idebenone]] || [https://clinicaltrials.gov/ct2/show/NCT01303406 NCT01303406] || 2011-2016 || EU || Friedreich Ataxia || 3 || 有効性なし
| |
| |-
| |
| | [[A0001]] ([[α-tocopherolquinone]]) || [https://clinicaltrials.gov/ct2/show/NCT01035671 NCT01035671] || 2009-2011 || 米国 || Friedreich Ataxia || 2 || [https://onlinelibrary.wiley.com/doi/full/10.1002/mds.25058 容量依存性にFRDA評価スケール改善]
| |
| |-
| |
| | [[Erythropoietin]] || [https://clinicaltrials.gov/ct2/show/NCT01016366 NCT01016366] || 2009-2016 || EU || Friedreich Ataxia || 2 || 有効性なし
| |
| |-
| |
| | idebenone || [https://clinicaltrials.gov/ct2/show/NCT00993967 NCT00993967] || 2009-2016 || EU || Friedreich Ataxia || 3 || 認容性を副作用の確認
| |
| |-
| |
| | [[deferiprone]] || [https://clinicaltrials.gov/ct2/show/NCT00530127 NCT00530127] || 2007-2009 || EU || Friedreich Ataxia || 1,2 || [https://onlinelibrary.wiley.com/doi/full/10.1002/ana.24248 非重症例で進行抑制の可能性(+)]
| |
| |-
| |
| | [[EGb 761]] || [https://clinicaltrials.gov/ct2/show/NCT00824512 NCT00824512] || 209-2013 || フランス || Friedreich Ataxia || 2 || 有効性なし
| |
| |-
| |
| | [[pioglitazone]] || [https://clinicaltrials.gov/ct2/show/NCT00811681 NCT00811681] || 2008-2013 || フランス || Friedreich Ataxia || 3 || 終了 | |
| |-
| |
| | [[varenicline ]]|| [https://www.clinicaltrials.gov/ct2/show/NCT00803868 NCT00803868] || 2008-2010 || 米国 || Friedreich Ataxia || 2,3 || 途中で中止
| |
| |-
| |
| | [[Triheptanoin]] || [https://clinicaltrials.gov/ct2/show/NCT04513002 NCT04513002] || 2020- || オーストラリア || Ataxia Telangiectasia || 2 || リクルート開始前
| |
| |-
| |
| | [[vitamin B3]] || [https://clinicaltrials.gov/ct2/show/NCT03962114 NCT03962114] || 2019- || オランダ || Ataxia Telangiectasia || 2 || リクルート中
| |
| |-
| |
| | [[N-Acetyl-L-Leucine]] || [https://clinicaltrials.gov/ct2/show/NCT03759678 NCT03759678] || 2018- || 米,英,独 || Ataxia Telangiectasia || 2 || リクルート中
| |
| |-
| |
| | [[Metformin]],[[Pioglitazone]] || [https://clinicaltrials.gov/ct2/show/NCT02733679 NCT02733679] || 2016-2019 || 英国 || Ataxia Telangiectasia || 4 || 終了
| |
| |-
| |
| | [[Somatropin]], [[Clonidine]], [[L-Arginin-Hydrochloride]], [[Estradiol valerate]] || [https://clinicaltrials.gov/ct2/show/NCT01052623 NCT01052623] || 2010-2011 || ドイツ || Ataxia Telangiectasia || 4 || 結果? | |
| |-
| |
| | [[amantadine sulphate]] || [https://clinicaltrials.gov/ct2/show/NCT00950196 NCT00950196] || 2009-2011 || イスラエル || Ataxia Telangiectasia || 4 || [https://journals.sagepub.com/doi/10.1177/0883073812441999 運動症状を改善]
| |
| |-
| |
| | [[Baclofen]] || [https://clinicaltrials.gov/ct2/show/NCT00640003 NCT00640003] || 2008-2011 || 米国 || Ataxia Telangiectasia || 1 || 結果?
| |
| |}
| |
|
| |
|
| == 疫学 == | | ===[[常染色体劣性遺伝]]性脊髄小脳変性症=== |
| 国や地域ごとに分布は異なっている。
| | ====概念==== |
| | 早期から緩徐進行性の小脳性運動失調を呈し、両親がいとこ婚である場合には、常染色体劣性遺伝性脊髄小脳変性症(autosomal recessive SCD:ARSCD)が疑われる。SCAと同じく、HUGOではSCARとして順番に番号がふられており、現在SCAR20まで登録されている(表3)。常染色体劣性遺伝性脊髄小脳変性症では純粋小脳型は少なく、末梢神経障害、眼球運動失行(ocular motor apraxia:OMA)などの多彩な症候を合併することが多い。 |
|
| |
|
| 弧発性脊髄小脳変性症では多系統萎縮症の頻度が最も高い。多系統萎縮症では、本邦では病初期に小脳症状が主体であるMSA-C (以前はオリーブ橋小脳萎縮症;olivopontocerebellar atrophy;OPCAと診断されていた臨床型) の頻度が高いが<ref name=Ozawa2010><pubmed>20571046</pubmed></ref> 、ヨーロッパや北米では病初期はパーキンソン症状が主体であるMSA-P (以前は線条体黒質変性症;striatonigral degeneration;SNDと診断されていた臨床型) の頻度が高い<ref name=Stefanova2009><pubmed>19909915</pubmed></ref> 。
| | ====各論==== |
| | #Friedreich運動失調症 Friedreich ataxia(FRDA)<br> 欧米では最も頻度が高い遺伝性脊髄小脳変性症である。Friedreich運動失調症の90%以上は、原因遺伝子[[frataxin]]のイントロンに存在するGAAリピートの著明な異常伸長のホモ接合体であり、数%は異常伸長と点変異の複合ヘテロ接合体である。しかし、欧米のFriedreich運動失調症には強い[[創始者効果]]が認められるため、わが国ではGAAリピートの異常伸長によるFriedreich運動失調症は確認されていない。原因遺伝子産物は、ミトコンドリアTCAサイクルを構成する[[aconitase]]などの[[鉄-硫黄タンパク質]]の機能維持に関与するので、Friedreich運動失調症の病態はfrataxinの機能喪失によるミトコンドリアの機能障害と想定される。<br> Friedreich運動失調症の主な症候は、[[後索]]の変性による[[深部感覚障害]]、錐体路症状、[[凹足]]、[[脊柱側弯症]]などである。小脳の萎縮は軽度であり、また[[心筋障害]]、[[糖尿病]]を合併する。 |
| | #アプラタキシンaprataxin欠損症<br> わが国では、眼球運動失行と[[wikipedia:ja:低アルブミン血症|低アルブミン血症]]という特異な症候を伴い、Friedreich運動失調症に類似した臨床像を呈する[[早発性失調症]]([[early onset ataxia with ocular motor apraxia and hypoalbuminemia]]/[[ataxia-ocular motor apraxia type 1]]:[[EAOH]]/[[AOA1]])が見出され、原因遺伝子としてaprataxinが同定された。GAAリピートの異常伸長を伴う欧米型のFriedreich運動失調症はわが国には存在しないと考えられるので、これまでわが国でFriedreich運動失調症として報告されてきた症例は本症と考えられ、本症はわが国の常染色体劣性遺伝性脊髄小脳変性症の約3分の2を占めている。原因遺伝子産物のaprataxinは[[核小体]]に局在するタンパク質であり、1本鎖DNAの損傷修復機構への関与が想定される。<br> 眼球運動失行では[[衝動性眼球運動]](saccade)の開始が著明に障害される。主に小児期に認められるため、本症は小児科領域でAOA1として記載されてきた。眼球運動失行は10代後半には次第に目立たなくなり、代わって眼球運動障害が進行してくる。また低アルブミン血症は30歳前後から明らかになる。 |
| | #セナタキシンsenataxin欠損症<br> Ataxia-ocular motor apraxiaには、AOA1に類似した臨床症状を呈しながら、アルブミンは低下せず、[[wikipedia:ja:α-fetoprotein|α-fetoprotein]]の高値を伴う[[AOA2]]がある。原因遺伝子[[senataxin]]の変異による。わが国からも報告があり、血中[[CK]]、[[wikipedia:ja:γ-グロブリン|γ-グロブリン]]も高値となる。 |
| | #サクシンsacsin欠損症<br> わが国の常染色体劣性遺伝性脊髄小脳変性症では、アプラタキシン欠損症に次いで、[[シャルルボア・サグネイ型劣性遺伝性痙性失調症]]([[autosomal recessive spastic ataxia of Charlevoix-Saguenay]]:[[ARSACS]];[[サクシン欠損症]])が多い。シャルルボア・サグネイ型劣性遺伝性痙性失調症は当初カナダのQuebec州から報告されたが、その後世界各地で見出されている。ケベックの症例は[[網膜有髄線維]]の増加を伴う痙性失調症を特徴とするが、わが国では網膜有髄線維を欠く例、痙縮を欠く例も報告されている。 |
| | #ビタミンE欠乏症<br> [[α-Tocopherol transfer protein]]の欠損による[[ビタミンE欠乏症]]では、進行性の小脳性運動失調が認められ、しばしば[[網膜]]色素変性を伴う。ビタミンEの大量投与により症状の改善が期待できるので、運動失調症の鑑別上重要である。 |
|
| |
|
| 遺伝性脊髄小脳変性症も、国や地域により疾患の分布は大きく異なる。世界規模では顕性遺伝のSCDでは、MJD (SCA3)の頻度が最も高く、それに続きSCA2 やSCA6の頻度が高いとされている。日本でもMJD、SCA6の頻度が高く、この他にSCA31の頻度が高い。また、日本国内においても、地域ごとに頻度が異なり、北海道や宮城県ではSCA1が他の地域と比較して頻度は高い。長野県や鳥取県では、MJDよりSCA6の頻度が高い<ref name=石川2015>'''石川欽也 (2015).'''<br>【小脳の最新知見-基礎研究と臨床の最前線】小脳の病態 小脳疾患の診療の最前線 日本に多い優性遺伝性脊髄小脳変性症(SCA3、6、31、DRPLA). 医学のあゆみ. 255, 1026-1032</ref> 。
| | ===遺伝性痙性対麻痺=== |
| | ====概念==== |
| | わが国の難治性疾患克服研究事業では、[[遺伝性痙性対麻痺]](Hereditary spastic paraplegia:HSP;spastic gait:SPG)が従来から脊髄小脳変性症に含まれており、脊髄小脳変性症全体の約4%を占めている。AD、AR、X染色体連鎖劣性の各遺伝形式をとるが、ADが多い。Hereditary spastic paraplegiaもspastic gaitの何番というように、病名を順番に機械的に決める方式が広く採用されており、その数は50を超えている(表4)。わが国では、ADでspastinの変異による[[SPG4]]が最も多い。 |
|
| |
|
| 潜性遺伝の脊髄小脳変性症では、白人ではFriedreich失調症の頻度が最も高いが、日本ではFriedreich失調症は1例も認められてない。一方日本では、Friedreich失調症に類似の臨床症状を示し、眼球運動失行、低アルブミン血症を合併するearly onset ataxia with ocular motor apraxia and hypoalbuminemia (EAOH) /ataxia-ocular motor apraxia type1 (AOA1) の頻度が高い<ref name=Yokoseki2011><pubmed>21486904</pubmed></ref> 。また、これまで日本からFriedreich失調症として報告されていた症例の多くが、EAOHであると考えられている。
| | ====症候==== |
| | HSPには、緩徐進行性の痙性対麻痺のみを呈する純粋型と、その他の症候を合併する複合型がある。複合型には小脳性運動失調を合併する場合があり、この場合は痙性対麻痺を主とする立場と、小脳性運動失調症を主体とする立場で分類が異なることになる。これまでにわが国で確認されている主な病型と原因遺伝子、臨床症状を表にまとめる。 |
|
| |
|
| ==関連項目==
| |
| *[[トリプレット病]]
| |
| ==参考文献== | | ==参考文献== |
| <references /> | | <references /> |
西澤 正豊
新潟大学 脳研究所
DOI:10.14931/bsd.6769 原稿受付日:2016年1月30日 原稿完成日:2016年月日
担当編集委員:漆谷 真(滋賀医科大学 医学部 神経内科)
英語名:spinocerebellar degeneration
英語略:SCD
概念
脊髄小脳変性症は、小脳あるいはその連絡線維の変性により、主な症状として小脳性運動失調を呈する疾患の総称である。
脊髄小脳変性症は従来、神経病理学的所見に基づいて、主に脊髄を障害するもの、脊髄と小脳を障害するもの、主に小脳を障害するものの3群に分類されてきた。しかし、最近では遺伝形式と臨床症候に基づく簡便な分類が用いられ、脊髄小脳変性症はまず孤発性と遺伝性に大別される。全体の約3分の2を占める孤発性群はさらに、変性が小脳に限局する皮質性小脳萎縮症(cortical cerebellar atrophy:CCA)と、変性が小脳系だけでなく、大脳基底核系や自律神経系、錐体路にも拡がる多系統萎縮症(multiple system atrophy:MSA)に分けられる。孤発性群では、多系統萎縮症が約3分の2、皮質性小脳萎縮症が約3分の1を占める。全体の残り3分の1は遺伝性群で、遺伝形式によって優性遺伝性と劣性遺伝性に分けられる。優性遺伝性が9割以上を占める。
遺伝性脊髄小脳変性症の原因遺伝子の同定が進み、分子病態が解明されつつある現状から、脊髄小脳変性症を病理学的な概念である「変性症」に限定せず、運動失調(ataxia)を呈する疾患群として捉えようとする立場や、分子病態に基づいて分類し直そうとする試みがある。
多系統萎縮症
概念
多系統萎縮症(Multiple system atrophy : MSA)の多系統変性は、小脳系、大脳基底核系、自律神経系の3系統を中心とし、錐体路にも及ぶ。小脳系の系統変性を主体とする病型は、従来、オリーブ橋小脳萎縮症(olivopontoserebellar atrophy:OPCA)、大脳基底核系では線条体黒質変性症(striatonigral degeneration:SND)、自律神経系ではShy-Drager症候群(Shy-Drager syndrome:SDS)と呼ばれてきた。
オリーブ橋小脳萎縮症はDejerineとAndré-Thomasによる1900年の報告に始まるが、オリーブ小脳系を超えた病変も認められていた。1964年にAdamsが提唱した線条体黒質変性症においても、黒質線条体だけでなく、オリーブ小脳系の変性を伴うと記載されていた。Shy-Drager症候群はShyとDragerにより1960年に報告されたが、1967年のSchwarzによる4剖検例では、自律神経系を超えた変性が認められていた。こうした経緯から、GrahamとOppenheimerは1969年、病変分布の共通性から、オリーブ橋小脳萎縮症、線条体黒質変性症、Shy-Drager症候群を包括する多系統委縮症という名称を提案した。高橋によるShy-Drager症候群のわが国初の詳細な剖検報告(1969年)でも、Shy-Drager症候群とオリーブ橋小脳萎縮症病変の共通性が指摘されている。
その後、多系統萎縮症に共通する疾患特異的バイオマーカーとして、脳幹のオリゴデンドログリアや神経細胞の細胞質内に特徴的な封入体(glial cytoplasmic inclusion:GCI、neuronal cytoplasmic inclusion:NCI)が見出され、多系統萎縮症は疾患単位として確立された。さらに、glial cytoplasmic inclusion、neuronal cytoplasmic inclusionの主な構成成分は、リン酸化されたα-シヌクレインであることが明らかにされた。
多系統萎縮症の診断には、1999年に発表されたGilmanらによるconsensus statementが広く用いられてきた。これによると、多系統萎縮症は診断の確かさによりdefinite、probable、possibleの3群に分類され、さらにオリーブ橋小脳萎縮症も線条体黒質変性症もいずれは自律神経症状を合併することからShy-Drager症候群を除外して、小脳症状と自律神経障害を呈して従来のオリーブ橋小脳萎縮症に相当する多系統萎縮症をMSA-C、パーキンソン症状と自律神経障害を呈して従来の線条体黒質変性症に相当する多系統萎縮症をMSA-Pとして、多系統萎縮症を臨床的に2分した。2008年には、改訂版が発表され、probableとpossibleの主な分岐点は、自律神経症状の程度により規定された。排尿障害では尿失禁、男性では勃起障害が重視され、起立性低血圧では、起立後3分以内に収縮期血圧が30 mmHg以上,あるいは拡張期血圧が15 mmHg以上低下する場合をprobableとする基準値が定められた(表1)。
これに対してわが国では、MSA-AとしてShy-Drager症候群を残そうとする立場もある。新潟大学脳研究所で、病理学的に診断が確定された多系統萎縮症の臨床像を検討すると、MCA-C、MSA-Pのいずれも22%は、初発症状が自律神経障害であった。Shy-Drager症候群とされてきた症例は、早期から著明な自律神経障害で発症し、次第に小脳性運動失調やパーキンソン症状を伴うが、Shy-Drager症候群に特異的な自律神経障害は指摘できない。また「premotor MSA」(発症早期に自律神経障害が前景に立ち、他の系統変性による症候がまだ目立たない段階で、たまたま病理学的検索が行われた症例)では、オリーブ橋小脳系と線条体黒質系の変性は軽微であるのに対し、脳幹の自律神経諸核には既にglial cytoplasmic inclusionを認めている。また、Shy-Drager症候群と進行性自律神経機能不全症(progressive autonomic failure:PAF)との鑑別も、初期には困難である。こうした知見を総合すると、Shy-Drager症候群を独立した疾患とすることは現時点では難しいと考えられる。
MSA-CとMSA-Pの頻度には、著明な人種差がある。わが国ではMSA-Cが全体の7、8割、MSA-Pが2、3割を占めるが、欧米ではこの頻度が逆転している。MSA-CとMSA-Pは臨床診断であるが、病理学的に診断が確定されたdefinite 多系統萎縮症についても、Wenningらが検討した欧州ではMSA-Pが8割を占め、一方、新潟大学の多系統萎縮症連続剖検例では、MSA-Cが3分の2を占めた。
病理所見としては、MSA-Cでは小脳皮質、橋小脳系、および下オリーブ核に強い変性と神経細胞脱落、グリオーシスが認められる。一方、MSA-Pでは被殻、黒質の変性が高度であり、特に被殻の後外側部は神経細胞脱落が強く、褐色調の色素沈着がみられる。Shy-Drager症候群とされた剖検例では、脊髄中間外側核、迷走神経背側核、交感神経節などの自律神経諸核の変性が強い。
glial cytoplasmic inclusionの主要な構成タンパク質であるα-シヌクレインは、もともとオリゴデンドログリアには発現していない。多系統萎縮症では病的グリアがα-シヌクレインを産生するという可能性よりも、神経細胞が産生したα-シヌクレインが細胞間を伝搬してグリアに取り込まれるという「プリオン様のタンパク伝搬仮説」が現在は有力である。パーキンソン病の特徴であるレヴィー小体の主な構成成分もリン酸化α-シヌクレインであるが、同じシヌクレイノパチーである多系統萎縮症とパーキンソン病がどこで分岐するかは未解明である。α-シヌクレイン遺伝子の点変異は家族性パーキンソン病の原因とはなるが、多系統萎縮症の表現型は示さない。α-シヌクレイン遺伝子のduplication、あるいはtriplicationによるまれな家族性パーキンソン病では、レヴィー小体とglial cytoplasmic inclusionがともに認められることから、遺伝子量の増大はglial cytoplasmic inclusion形成の原因の一つと考えられる。
ごくまれではあるが、多系統萎縮症には家族発症例があり、これらの解析から辻らによりCOQ2(コエンザイムQ10合成酵素)遺伝子に変異が同定された。変異が2つあれば発症者となり、変異が1つでは発症リスクを高めることになる。日本人のみに認められるV393A変異は多系統萎縮症の約9%に見出され(健常者では約3%)、ホモ変異例では脳内のコエンザイムQ10量が減少していた。
表1.多系統萎縮症診断基準改訂版[1]
従来通り、definite, probable, possibleに分類し、さらにMSA-PとMSA-Cに分類する。
- Definite MSA
病理学的に,中枢神経に広範に、多数のα-synuclein陽性glial cytoplasmic inclusion(GCI)を認め、線条体黒質系またはオリーブ橋小脳系の変性所見を伴う。
- Probable MSA
孤発性で進行性の成人発症(30歳以降)の変性疾患で、自律神経障害(尿失禁(膀胱からの尿排出をコントロールできない、男性では勃起障害)、または起立後3分以内に少なくとも収縮期血圧が30 mmHg,拡張期血圧が15 mmHg低下する起立性低血圧)に加え、レボドパ反応性の乏しいパーキンソニズム(動作緩慢に、筋強剛、振戦、または姿勢反射障害を伴う)、または小脳症候群(歩行失調に、小脳性構音障害、四肢失調、または小脳性眼球運動障害を伴う)を呈する。
- Possible MSA
孤発性で進行性の成人発症(30歳以降)の変性疾患で、パーキンソニズム、または小脳症候群を呈し、加えて自律神経障害を示唆する所見(他の原因では説明できない尿意促迫、頻尿、残尿、男性では勃起不全、またはprobable MSAの規準を満たさないレベルの起立性低血圧)を少なくとも一つ認め、さらに以下の表で少なくとも一つの所見を満たすもの。
(1) Possible MSA-P またはMSA-C
腱反射亢進を伴うBabinski徴候陽性、喘鳴。
(2) Possible MSA-P
急速進行性のパーキンソニズム、レボドパ反応性が乏しいこと、運動症状出現3年以内の姿勢反射障害、
歩行失調・小脳性構音障害・四肢失調・または
小脳性眼球運動障害、運動症状出現5年以内の嚥下障害、
MRIにおける被殻・中小脳脚・橋・または小脳の萎縮、FDG-PETにおける被殻・脳幹・または小脳の低代謝。
(3) Possible MSA-C
パーキンソニズム(動作緩慢と筋強剛)、MRIにおける被殻・中小脳脚・または橋の萎縮、FDG-PETにおける被殻の低代謝、SPECTまたはPETにおける
黒質線条体ドーパミン作動性ニューロンの節前性脱神経 。
|
多系統萎縮症の診断を支持するred flag所見
口部顔面ジストニア、頸部前屈、カンプトコルミア(脊柱の高度の前屈)and/or Pisa症候群(脊柱の高度の側屈)、手または足の拘縮、吸気時のため息、高度の発声困難、高度の構音障害、いびきの出現または増悪、手足の冷感、病的笑いまたは病的泣き、jerkyなミオクローヌス様の姿勢振戦または動作性振戦。
|
多系統萎縮症の診断を支持しない所見
典型的丸薬丸め様の静止時振戦、臨床的に有意な末梢神経障害、薬剤誘発性でない幻覚、75歳以上の発症、失調症やパーキンソニズムの家族歴、認知症(DSM-IVによる)、多発性硬化症を示唆する大脳白質病変。
|
症候
MSA-Cは40~60歳に、多くは小脳性運動失調から発症し、次第に自律神経症状や錐体外路症状、錐体路症状を伴う。新潟大学の剖検例では、MSA-Cにパーキンソニズムを伴うのは74%であった。また、尿失禁や排尿困難、起立性低血圧や失神、男性では陰萎などの自律神経症状が発現する中央値は発症から2.5年であり、2.5年より早期から自律神経障害が出現すると、その後の進行が速かった。
MSA-Pの多くはパーキンソン症状から発症し、次第に自律神経症状を伴う。小脳性運動失調症状はパーキンソン症状にマスクされやすく、MSA-Pが小脳性運動失調を伴う頻度は、新潟大学の検討では44%であった。MSA-Pの初期には、パーキンソン病との鑑別が困難な症例もある。パーキンソン病に比べて、レボドパ補充療法の効果が乏しく、進行が速く、症状の左右差や静止時振戦がまれであることが特徴とされるが、MSA-Pでもパーキンソン症状の左右差が明らかな例や、典型的な静止時振戦を示す例、レボドパも無効ではなく、改善を示す例がある。進行期になると、多系統萎縮症でも大脳皮質の著明な萎縮や、進行性の認知障害が認められる。
多系統萎縮症の全経過は約9年で、誤嚥性肺炎や敗血症などの感染症が死因となることが多いが、夜間の突然死も重要である。通常の低音のいびきとは異なる高調の喉頭喘鳴は、声帯外転麻痺を示唆する症候とされ、声帯外転麻痺による気道閉塞が突然死の原因と考えられてきた。しかし、麻酔薬により睡眠状態を再現して喉頭内視鏡検査を行うと、気道狭窄が生じている部位は声帯に限らず、被裂部、喉頭蓋、舌根部、軟口蓋など広範囲に及び、また吸気時に喉頭蓋が気管に引き込まれ、気道を閉塞するfloppy epiglottisと呼ばれる病態も合併することが明らかになった。MSAの睡眠呼吸障害に対する治療法として、マスクを用いた持続陽圧換気(continuous positive airway pressure: CPAP)を不用意に行うと、floppy epiglottisでは気道狭窄が悪化する恐れがあり、注意を要する。
多系統萎縮症の睡眠呼吸障害に対して、CPAP装着や気管切開などを行っても、突然死を防げない症例が存在する。中枢性無呼吸や致死性不整脈などが原因と考えられ、気管切開による人工呼吸管理が必要になる。
補助診断法
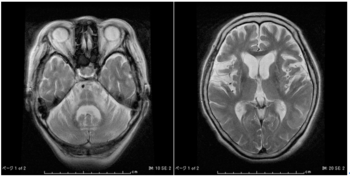
図1.多系統萎縮症のMRI所見図左:MSA-Cにおける橋十字サインと橋、小脳の萎縮
図右:MSA-Pにおける線条体後外側部の線状高信号(スリットサイン)
多系統萎縮症の補助診断にはMRIが有用である。MSA-Cでは、小脳、中小脳脚、脳幹の進行性萎縮とともに、橋底部に十字状の高信号(hot cross bun sign:橋十字サイン)が、MSA-Pでは、被殻の進行性萎縮とグリオーシス、鉄の沈着により、被殻後外側部に線状の高信号(putaminal slit sign)が認められる(図1)。
MIBG心筋シンチグラフィーでは、MSA-Pの初期には取り込みの低下は認められないので、パーキンソン病との鑑別に役立つ。脳脊髄液中のα-シヌクレインは多系統萎縮症では低下する。glial cytoplasmic inclusionに結合するリガンドを利用したPET検査も開発中である。
治療
根治的治療法は確立されておらず、対症療法が主体となる。わが国では、thyrotropin releasing hormone(TRH)の点滴とその誘導体(タルチレリン)の経口投与が、小脳性運動失調に対して唯一保険適用となっているが、その効果は限定的である。起立性低血圧や排尿障害などの自律神経症状には、対症療法を行う。多くの薬剤について、小脳性運動失調症に対する有効性が検証されているが、確実に効果が実証されたものはない。
多系統萎縮症では、経過中に気道や尿路の感染症を繰り返して、全身状態が悪化することが多い。口腔ケアを徹底して、誤嚥による気道感染を予防することが重要である。
脊髄小脳変性症と多系統萎縮症は厚生労働省の指定難病制度の対象疾患であり、さらに介護保健法における「特定疾病」に指定されている。制度上Shy-Drager症候群を拡大して多系統萎縮症として独立させたために、脊髄小脳変性症には皮質性小脳萎縮症と遺伝性脊髄小脳変性症が残された形となっている。また、MSA-Pはパーキンソン病と診断されている場合が少なからずあり、難病対策制度上の分類には、再度整理が必要である。
皮質性小脳萎縮症
概念
脊髄小脳変性症の中では最も高齢で発症し、小脳性運動失調のみが緩徐に進行する孤発性の一群を皮質性小脳萎縮症(Cortical cerebellar atrophy : CCA)と呼んでいる。しかし、皮質性小脳萎縮症は単一疾患ではなく、一見家族歴を欠いていても、遺伝子診断により後述するSCA6やSCA31と確定される例があり、またアルコール性などの二次性小脳変性症も含まれる。純粋小脳型を呈する変性疾患としての皮質性小脳萎縮症は、実際には非常に少ないと考えられる。
症候
中年期以降に、小脳性の体幹運動失調と構音障害が緩徐に進行する。経過は多系統萎縮症に比べて緩やかであり、進行しても独立歩行が可能な例もある。四肢の協調運動障害も次第に進行するが、小脳系以外の症候は認めない。
補助診断法
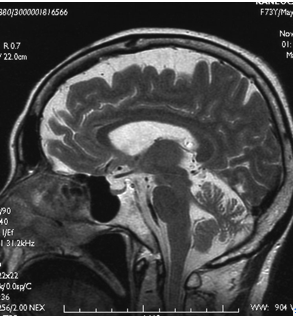
図2.皮質性小脳萎縮症のMRIにおける小脳萎縮小脳の萎縮を認めるが、脳幹は保たれている
画像検査では、小脳に限局して進行性の萎縮を認める(図2)。病初期には虫部前葉から萎縮が始まり、次第に小脳半球に波及する。しかし、甲状腺機能低下症、ビタミンE欠乏症、ビタミンB1欠乏症、Wilson病などの代謝性疾患、慢性アルコール中毒、フェニトインや臭化バレリル尿素などの薬物中毒、有機水銀中毒、トルエンやベンゼンなどの有機溶媒中毒、傍腫瘍性小脳変性症(腫瘍随伴性神経症候群)、グルテン失調症、GAD抗体陽性失調症、急性小脳炎、Fisher症候群、神経Behçet病、多発性硬化症、小脳血管障害、小脳腫瘍など、多くの疾患を除外する必要があり、診断を皮質性小脳萎縮症と確定することは容易ではない。
治療
根治的な治療法は確立されていないが、小脳の機能維持を目的として、四肢末梢への錘負荷やバランス訓練などのリハビリテーションが広く行われてきた。小脳が正常に保たれている脳血管障害に対する機能回復訓練とは異なり、運動学習の首座と考えられる小脳に進行性の変性が起きている小脳変性症の場合にも、繰り返し学習による可塑性(use- dependent plasticity)が獲得されるか否かは明らかでなかった。そこで、厚生労働省の運動失調症調査研究班で筆者らは、短期集中リハビリが小脳性運動失調の進行抑制に有効であるかを検証する臨床治験を、皮質性小脳萎縮症と遺伝性純粋小脳型失調症(SCA6とSCA31)を対象として実施し、1日各1時間の理学療法と作業療法を1ヶ月間継続すると、小脳性運動失調は改善し、その効果は最大6ヶ月続くことが実証された。この効果は既存の薬物治療効果を上回っており、小脳機能維持を目的としたリハビリテーション体制を整備することが今後の課題である。
遺伝性脊髄小脳変性症
常染色体優性遺伝性脊髄小脳変性症
概念
遺伝性脊髄小脳変性症の9割以上を占める常染色体優性遺伝性脊髄小脳変性症(Autosomal dominant SCD:ADSCD)は、その約9割まで原因遺伝子が同定された。原因遺伝子座が同定された常染色体優性遺伝性脊髄小脳変性症は、脊髄小脳失調症(spinocerebellar ataxia:SCA)の何番というように、病名を機械的に決める方式が広く採用されている。The Human Genome Organization(HUGO)には現在SCA41まで登録されており、このうちSCA9、16、22は欠番である。一方、わが国で頻度が高い歯状核赤核淡蒼球ルイ体萎縮症(dentatorubral pallidoluysian atrophy:DRPLA)は、脊髄小脳失調症としては登録されていない。
わが国ではMachado-Joseph病(MJD:別名SCA3)の頻度が最も高く、全体の約4分の1を占める。SCA6、DRPLA、SCA31がこれに次ぐ。これらの頻度には地域差があり、東日本ではMachado-Joseph病、西日本ではSCA6が多い。
常染色体優性遺伝性脊髄小脳変性症における遺伝子異常の多くは、翻訳領域に存在するCAGリピート長が正常の2、3倍に異常伸長していることであり、遺伝子レベルではCAGリピート病、タンパク質レベルではポリグルタミン病とよばれる。伸長したポリグルタミン鎖を含むタンパク質が凝集する過程で形成されるオリゴマーに細胞障害性があると考えられる。
ポリグルタミン病では、世代を経る毎に発症年齢が若年化し、重症化する表現促進現象(anticipation)が認められる。Mendel遺伝では説明できない現象であったが、リピート数の伸長によることが明らかになっている。翻訳領域のCAGリピートは父方から伝搬する場合に著明に伸長する傾向があり、CAGリピート数が短いSCA6を除き、発症年齢とリピート数には負の相関が認められる。
遺伝性脊髄小脳変性症に関する遺伝子診断を行う際には、文部科学省、厚生労働省、経済産業省の3省庁合同のヒトゲノム・遺伝子解析研究に関する最新の倫理指針を遵守する必要がある。根治的な治療法が確立されていない遺伝性疾患の発症前診断や保因者診断は、原則として行わない。
各論
わが国で頻度の高い病型を中心とし、その他の病型は表2に一括した。
- Machado-Joseph病 MJD(SCA3)
Machado-Joseph病は当初、ポルトガル領アゾレス諸島から北米に移民した子孫の間に見出された疾患であり、その後、欧州で記載されたSCA3でも同一のCAGリピート伸長が確認されている。臨床的にはRosenbergにより、若年発症で錐体路症状と、ジストニアなどの錐体外路症状が目立つ1型、成年発症で痙性失調症と眼振を呈する2型、高齢発症で筋萎縮や末梢神経障害などの末梢性病変を伴う3型、パーキンソニズムを伴うまれな4型に分けられている。Ataxin3遺伝子に存在するCAGリピートの伸長は1型で最も長く、3型では短い。顔面筋の線維束性収縮やミオキミア、びっくり眼などはMachado-Joseph病によくみられる。
- SCA6
50歳前後で発症し、小脳性運動失調症状のみを呈する純粋小脳型常染色体優性遺伝性脊髄小脳変性症であり、P/Q型電位依存性Caチャネルα1サブユニット遺伝子のC末端に位置するCAGリピートの軽度の伸長による。同遺伝子の点変異は、反復発作性運動失調症2型(episodic ataxia type 2: EA2)と家族性片麻痺性片頭痛の原因でもある。
- SCA31
常染色体優性遺伝性脊髄小脳変性症では最も高齢の60歳前後で発症する純粋小脳型常染色体優性遺伝性脊髄小脳変性症であるが、遺伝子診断によらずにSCA6と鑑別することは困難である。わが国では長野県、静岡県、鹿児島県で特に多い。第16染色体長腕のBEANとTK2遺伝子に共通するイントロンに挿入されたTGGAAという5塩基リピートが著明に伸長しており、転写産物によるRNA fociが形成されていることから、これと相互作用する核タンパク質の機能変化が想定される。
- DRPLA
わが国に多い常染色体優性遺伝性脊髄小脳変性症で、発症年齢により臨床症状が異なる。atrophin 1遺伝子に存在するCAGリピートが長い場合は若年発症となり、進行性ミオクローヌスてんかんの臨床像を示す。伸長の程度が軽い場合には成人発症となり、認知機能障害や不随意運動などを呈する。ポリグルタミン病では最も著明な表現促進現象がみられ、リピート伸長の程度により、発症年齢や臨床像、重症度が規定される。小脳歯状核とその遠心路、淡蒼球視床下核系に変性と萎縮を認めるだけでなく、大脳白質にも広範な変性像が認められる。
- 毛細血管拡張運動失調症(ataxia telangiectasia:AT;Louis-Bar症候群)
幼児期に小脳性運動失調と皮膚や眼球結膜の毛細血管拡張症で発症する。IgAが低下し、免疫不全のために感染症を起こしやすく、また高率に悪性リンパ腫などの悪性腫瘍を合併する。ATの責任遺伝子ATMは2本鎖DNAの損傷修復に関与するタンパク質をコードする。神経症状として眼球運動失行を認め、以下に述べるaprataxinやsenataxinの欠損症と病態、臨床症候は類似している。
概念
早期から緩徐進行性の小脳性運動失調を呈し、両親がいとこ婚である場合には、常染色体劣性遺伝性脊髄小脳変性症(autosomal recessive SCD:ARSCD)が疑われる。SCAと同じく、HUGOではSCARとして順番に番号がふられており、現在SCAR20まで登録されている(表3)。常染色体劣性遺伝性脊髄小脳変性症では純粋小脳型は少なく、末梢神経障害、眼球運動失行(ocular motor apraxia:OMA)などの多彩な症候を合併することが多い。
各論
- Friedreich運動失調症 Friedreich ataxia(FRDA)
欧米では最も頻度が高い遺伝性脊髄小脳変性症である。Friedreich運動失調症の90%以上は、原因遺伝子frataxinのイントロンに存在するGAAリピートの著明な異常伸長のホモ接合体であり、数%は異常伸長と点変異の複合ヘテロ接合体である。しかし、欧米のFriedreich運動失調症には強い創始者効果が認められるため、わが国ではGAAリピートの異常伸長によるFriedreich運動失調症は確認されていない。原因遺伝子産物は、ミトコンドリアTCAサイクルを構成するaconitaseなどの鉄-硫黄タンパク質の機能維持に関与するので、Friedreich運動失調症の病態はfrataxinの機能喪失によるミトコンドリアの機能障害と想定される。
Friedreich運動失調症の主な症候は、後索の変性による深部感覚障害、錐体路症状、凹足、脊柱側弯症などである。小脳の萎縮は軽度であり、また心筋障害、糖尿病を合併する。
- アプラタキシンaprataxin欠損症
わが国では、眼球運動失行と低アルブミン血症という特異な症候を伴い、Friedreich運動失調症に類似した臨床像を呈する早発性失調症(early onset ataxia with ocular motor apraxia and hypoalbuminemia/ataxia-ocular motor apraxia type 1:EAOH/AOA1)が見出され、原因遺伝子としてaprataxinが同定された。GAAリピートの異常伸長を伴う欧米型のFriedreich運動失調症はわが国には存在しないと考えられるので、これまでわが国でFriedreich運動失調症として報告されてきた症例は本症と考えられ、本症はわが国の常染色体劣性遺伝性脊髄小脳変性症の約3分の2を占めている。原因遺伝子産物のaprataxinは核小体に局在するタンパク質であり、1本鎖DNAの損傷修復機構への関与が想定される。
眼球運動失行では衝動性眼球運動(saccade)の開始が著明に障害される。主に小児期に認められるため、本症は小児科領域でAOA1として記載されてきた。眼球運動失行は10代後半には次第に目立たなくなり、代わって眼球運動障害が進行してくる。また低アルブミン血症は30歳前後から明らかになる。
- セナタキシンsenataxin欠損症
Ataxia-ocular motor apraxiaには、AOA1に類似した臨床症状を呈しながら、アルブミンは低下せず、α-fetoproteinの高値を伴うAOA2がある。原因遺伝子senataxinの変異による。わが国からも報告があり、血中CK、γ-グロブリンも高値となる。
- サクシンsacsin欠損症
わが国の常染色体劣性遺伝性脊髄小脳変性症では、アプラタキシン欠損症に次いで、シャルルボア・サグネイ型劣性遺伝性痙性失調症(autosomal recessive spastic ataxia of Charlevoix-Saguenay:ARSACS;サクシン欠損症)が多い。シャルルボア・サグネイ型劣性遺伝性痙性失調症は当初カナダのQuebec州から報告されたが、その後世界各地で見出されている。ケベックの症例は網膜有髄線維の増加を伴う痙性失調症を特徴とするが、わが国では網膜有髄線維を欠く例、痙縮を欠く例も報告されている。
- ビタミンE欠乏症
α-Tocopherol transfer proteinの欠損によるビタミンE欠乏症では、進行性の小脳性運動失調が認められ、しばしば網膜色素変性を伴う。ビタミンEの大量投与により症状の改善が期待できるので、運動失調症の鑑別上重要である。
遺伝性痙性対麻痺
概念
わが国の難治性疾患克服研究事業では、遺伝性痙性対麻痺(Hereditary spastic paraplegia:HSP;spastic gait:SPG)が従来から脊髄小脳変性症に含まれており、脊髄小脳変性症全体の約4%を占めている。AD、AR、X染色体連鎖劣性の各遺伝形式をとるが、ADが多い。Hereditary spastic paraplegiaもspastic gaitの何番というように、病名を順番に機械的に決める方式が広く採用されており、その数は50を超えている(表4)。わが国では、ADでspastinの変異によるSPG4が最も多い。
症候
HSPには、緩徐進行性の痙性対麻痺のみを呈する純粋型と、その他の症候を合併する複合型がある。複合型には小脳性運動失調を合併する場合があり、この場合は痙性対麻痺を主とする立場と、小脳性運動失調症を主体とする立場で分類が異なることになる。これまでにわが国で確認されている主な病型と原因遺伝子、臨床症状を表にまとめる。
参考文献
- ↑
Gilman, S., Wenning, G.K., Low, P.A., Brooks, D.J., Mathias, C.J., Trojanowski, J.Q., ..., & Vidailhet, M. (2008).
Second consensus statement on the diagnosis of multiple system atrophy. Neurology, 71(9), 670-6.
[PubMed:18725592]
[PMC]
[WorldCat]
[DOI]

