「アミロイドーシス」の版間の差分
細編集の要約なし |
Makotourushitani (トーク | 投稿記録) 細編集の要約なし |
||
| (4人の利用者による、間の9版が非表示) | |||
| 2行目: | 2行目: | ||
<font size="+1">[http://researchmap.jp/_tomitataisuke 富田 泰輔]</font><br> | <font size="+1">[http://researchmap.jp/_tomitataisuke 富田 泰輔]</font><br> | ||
''東京大学 薬学研究科''<br> | ''東京大学 薬学研究科''<br> | ||
DOI [[XXXX]]/XXXX 原稿受付日:2013年11月27日 原稿完成日:2013年月日<br> | |||
担当編集委員:[http://researchmap.jp/read0141446 漆谷 真] | 担当編集委員:[http://researchmap.jp/read0141446 漆谷 真](京都大学 大学院医学研究科)<br> | ||
</div> | </div> | ||
英:amyloidosis | |||
{{box|text= [[アミロイド]]amyloidは[[wikipedia:ja:コンゴーレッド|コンゴーレッド]]染色でオレンジ色に染まり、[[wikipedia:ja:偏光顕微鏡|偏光顕微鏡]]で緑色偏光を呈し、[[wikipedia:ja:電子顕微鏡|電子顕微鏡]]観察下では7~15nmの繊維構造を呈する物質である。アミロイドが、組織間隙に沈着して臓器の機能不全が生じる疾患をアミロイドーシス amyloidosisと呼ぶ<ref><pubmed> 22664198 </pubmed></ref>。アミロイドタンパク質の種類や臓器によって特徴が見られ、大きく[[全身性アミロイドーシス]]と[[限局性アミロイドーシス]]に分類される。代表的な全身性アミロイドーシスには、全身性AAアミロイドーシス、家族性アミロイドニューロパチーが挙げられる。限局性アミロイドーシスには脳アミロイドーシスである、[[アルツハイマー病]]、[[脳血管アミロイドアンギオパチー]]、遺伝性アミロイド性脳出血で、[[クロイツフェルト・ヤコブ病]]などが知られている。基本的には、アミロイドーシス発症の分子病態は凝集するアミロイドタンパク質の濃度上昇か、凝集能亢進によるものである。したがってアミロイドタンパク質の除去が根本治療戦略となる。}} | {{box|text= [[アミロイド]]amyloidは[[wikipedia:ja:コンゴーレッド|コンゴーレッド]]染色でオレンジ色に染まり、[[wikipedia:ja:偏光顕微鏡|偏光顕微鏡]]で緑色偏光を呈し、[[wikipedia:ja:電子顕微鏡|電子顕微鏡]]観察下では7~15nmの繊維構造を呈する物質である。アミロイドが、組織間隙に沈着して臓器の機能不全が生じる疾患をアミロイドーシス amyloidosisと呼ぶ<ref><pubmed> 22664198 </pubmed></ref>。アミロイドタンパク質の種類や臓器によって特徴が見られ、大きく[[全身性アミロイドーシス]]と[[限局性アミロイドーシス]]に分類される。代表的な全身性アミロイドーシスには、全身性AAアミロイドーシス、家族性アミロイドニューロパチーが挙げられる。限局性アミロイドーシスには脳アミロイドーシスである、[[アルツハイマー病]]、[[脳血管アミロイドアンギオパチー]]、遺伝性アミロイド性脳出血で、[[クロイツフェルト・ヤコブ病]]などが知られている。基本的には、アミロイドーシス発症の分子病態は凝集するアミロイドタンパク質の濃度上昇か、凝集能亢進によるものである。したがってアミロイドタンパク質の除去が根本治療戦略となる。}} | ||
==アミロイドーシスとは== | ==アミロイドーシスとは== | ||
アミロイドamyloidはコンゴーレッド染色でオレンジ色に染まり、偏光顕微鏡で緑色偏光を呈し、電子顕微鏡観察下では7~15nmの繊維構造を呈する物質として定義される。アミロイドが、組織間隙に沈着する疾患を総称してアミロイドーシス amyloidosisと呼ぶ<ref><pubmed> 22664198 </pubmed></ref>。多くの場合、前駆タンパクであるアミロイドタンパク質が折りたたみ障害を引き起こして重合し、[[wikipedia:ja:βシート|βシート]]構造に富む不溶性線維として蓄積・凝集している | |||
沈着するアミロイドタンパク質の種類や臓器によって特徴が見られ、特に大きく全身性アミロイドーシスと限局性アミロイドーシスに分類されている。 | |||
===全身性アミロイドーシス=== | ===全身性アミロイドーシス=== | ||
| 37行目: | 37行目: | ||
{| class="wikitable" | {| class="wikitable" | ||
|- class="hintergrundfarbe5" | |- class="hintergrundfarbe5" | ||
! タイプ | ! タイプ | ||
! 頻度 | ! 頻度 | ||
! 詳細 | ! 詳細 | ||
|- | |- | ||
| ''' | | '''原発性アミロイドーシス''' || 稀 || 基礎疾患を伴わないアミロイドーシス | ||
|- | |- | ||
| ''' | | '''家族性アミロイドーシス''' || 稀 || 遺伝的に変異をおこしたアミロイド原性タンパク質によって惹起されるアミロイドーシス | ||
|- | |- | ||
| ''' | | '''二次性アミロイドーシス''' || 低 || 何らかの基礎疾患に合併して生じるアミロイドーシス<br /> 多発性骨髄腫や原発性マクログロブリン血症とALアミロイドーシス<br /> 関節リウマチとAAアミロイドーシス<br /> 長期透析とAβ<sub>2</sub>Mアミロイドーシス | ||
|- | |- | ||
| ''' | | '''老人性全身アミロイドーシス''' || 高 || 加齢とともに心臓を含めた全身においてトランスサイレチンが蓄積する<br /> 遺伝子変異を伴わず、野生型アミロイドタンパク質が蓄積する | ||
|- | |- | ||
|} | |} | ||
{| class="wikitable" class="sortable wikitable" | {| class="wikitable" class="sortable wikitable" | ||
|- | |- | ||
! 略称 | ! 略称 | ||
| 61行目: | 59行目: | ||
| '''AL''' | | '''AL''' | ||
| [[免疫グロブリンL鎖]] | | [[免疫グロブリンL鎖]] | ||
| | | 異常形質細胞によって産出されるモノクローナル免疫グロブリンL鎖由来のアミロイドALが全身諸臓器(心臓、腎臓、消化管、肝臓、末梢神経など)に沈着する。 | ||
| {{OMIM2|254500}} | | {{OMIM2|254500}} | ||
|- | |- | ||
| '''AA''' | | '''AA''' | ||
| [[血清アミロイドA]] | | [[血清アミロイドA]] | ||
| | | 慢性炎症時におもに肝臓から産出される急性期タンパク質の血清アミロイドA(SAA)の代謝産物アミロイドA(AA)が腎臓や消化管に沈着する。 | ||
|- | |- | ||
| '''Aβ''' | | '''Aβ''' | ||
| 75行目: | 73行目: | ||
| '''ATTR''' | | '''ATTR''' | ||
| [[トランスサイレチン]] | | [[トランスサイレチン]] | ||
| | | 主として肝臓から産生されるが、トランスサイレチン遺伝子に変異のある異型TTRは肝実質にアミロイド沈着はほとんどなく、神経節を含む末梢神経、自律神経系やほかの組織に沈着する。<br />野生型トランスサイレチンが蓄積する老人性全身性アミロイドーシスでは主に心臓、他に肺、腎臓、全身の小血管にアミロイドが蓄積する。 | ||
| {{OMIM2|105210}} | | {{OMIM2|105210}} | ||
|- | |- | ||
| '''Aβ<sub>2</sub>M''' | | '''Aβ<sub>2</sub>M''' | ||
| [[β2ミクログロブリン]] | | [[β2ミクログロブリン]] | ||
| | | 長期透析患者では血中で増加しているβ2ミクログロブリン由来のアミロイドが靱帯、骨領域に沈着する。 | ||
|- | |- | ||
| '''AIAPP''' | | '''AIAPP''' | ||
| [[アミリン]] | | [[アミリン]] | ||
| | | Ⅱ型糖尿病に伴い膵ランゲルハンス島やインスリノーマにアミリン由来のアミロイドが蓄積する。 | ||
|- | |- | ||
| '''APrP''' | | '''APrP''' | ||
| 128行目: | 126行目: | ||
| '''APro''' | | '''APro''' | ||
| [[プロラクチン]] | | [[プロラクチン]] | ||
| | | 下垂体のプロラクチン産生腺腫において見出されるアミロイドーシス。 | ||
| | | | ||
|- | |- | ||
| '''AKer''' | | '''AKer''' | ||
| [[ケラチン(ケラトエピセリン)]] | | [[ケラチン(ケラトエピセリン)]] | ||
| | | 皮膚に限局して発症するアミロイドーシス。 | ||
| | | | ||
|- | |- | ||
| '''AANF''' | | '''AANF''' | ||
| [[心房性ナトリウム利尿ペプチド]] | | [[心房性ナトリウム利尿ペプチド]] | ||
| | | 心房に限局して沈着するアミロイドーシス。 | ||
| | | | ||
|- | |- | ||
| '''ACal''' | | '''ACal''' | ||
| [[カルシトニン]] | | [[カルシトニン]] | ||
| | | 甲状腺髄様癌において見出されるアミロイドーシス。 | ||
| | | | ||
|} | |} | ||
==病態生理== | ==病態生理== | ||
[[Image:2M5N.pdb|thumb|350px|''' | [[Image:2M5N.pdb|thumb|350px|'''図1.クロスβ構造'''<br>トランスサイレチン部分ペプチドからなるクロスβ構造。PDB ID: {{PDB2|2M5N}}]] | ||
(編集コメント:結晶構造は回転できる物に取り替えました。これで良いかご確認下さい。SafariではWebGLをonにして下さい。) | |||
===構造=== | ===構造=== | ||
各アミロイドタンパク質には一定の共通したアミノ酸配列や構造は見られないが、アミロイド線維になると共通して[[クロスβ構造]]と呼ばれる形態をとっている<ref><pubmed> 17468747 </pubmed></ref><ref><pubmed> 21456964 </pubmed></ref><ref><pubmed> 23513222 </pubmed></ref>。これはアミロイド線維を構成するポリペプチド鎖が線維軸と垂直方向に[[wikipedia:ja:βストランド|βストランド]]となり、かつ線維軸方向に[[wikipedia:ja:βシート構造|βシート構造]] | 各アミロイドタンパク質には一定の共通したアミノ酸配列や構造は見られないが、アミロイド線維になると共通して[[クロスβ構造]]と呼ばれる形態をとっている<ref><pubmed> 17468747 </pubmed></ref><ref><pubmed> 21456964 </pubmed></ref><ref><pubmed> 23513222 </pubmed></ref>。これはアミロイド線維を構成するポリペプチド鎖が線維軸と垂直方向に[[wikipedia:ja:βストランド|βストランド]]となり、かつ線維軸方向に[[wikipedia:ja:βシート構造|βシート構造]]をとっているものである。このような構造学的特徴はイメージング技術に応用されつつあり、[[wikipedia:ja:Aβ|Aβ]]線維に特異的に結合する低分子化合物を利用した[[アミロイドPETスキャン]]が可能となった<ref><pubmed> 14991808 </pubmed></ref><ref><pubmed> 21245183 </pubmed></ref>。 | ||
===線維形成過程と伝播=== | ===線維形成過程と伝播=== | ||
[[Image:TTfig6.png|thumb|350px|''' | [[Image:TTfig6.png|thumb|350px|'''図2.アミロイド線維形成過程'''<br>アミロイド線維形成過程におけるシードの役割]] | ||
[[Image:2M4J.pdb|thumb|350px|''' | [[Image:2M4J.pdb|thumb|350px|'''図3.アルツハイマー病患者脳由来のAβ線維構造'''<br>患者脳由来アミロイドから伸長した[[アミロイドβタンパク質]]の分子構造。PDB ID: {{PDB2|2M4J}}]] | ||
アミロイド線維形成過程では、多くの場合正常なフォールディングをうけているアミロイドタンパク質が何らかの理由で一旦部分変性し、会合することが必要である。また線維形成過程はその鋳型となるシード(種、核)の形成を契機として急速に進んでいくことが示されている<ref><pubmed> 22885025 </pubmed></ref> | |||
(編集コメント:長かったので、小見出しをつけました。内容に照らして適当かご確認下さい。) | |||
アミロイド線維形成過程では、多くの場合正常なフォールディングをうけているアミロイドタンパク質が何らかの理由で一旦部分変性し、会合することが必要である。また線維形成過程はその鋳型となるシード(種、核)の形成を契機として急速に進んでいくことが示されている<ref><pubmed> 22885025 </pubmed></ref>。すなわち、このシードの両端の末端にアミロイドタンパク質が結合して線維が伸長していくと考えられている。 | |||
このようなシード依存性伸長反応モデルは、[[プリオン]]タンパク質が示す伝播能力とも関連していると考えられている。すなわち、一旦異常構造をとったタンパク質がシードとなり、別の個体におけるアミロイドタンパク質の構造及び性質を変化させていくというモデルである<ref><pubmed> 8513491 </pubmed></ref>。またシードへの組み込みはアミロイドタンパク質が同様の構造を取りうるかどうかに依存する。プリオンの感染性にはごく僅かなアミノ酸の違いに起因する「種の壁」が存在するが、この現象も一次配列の違いに依存する各種のプリオンが形成するシード構造の違いによって説明できる。 | このようなシード依存性伸長反応モデルは、[[プリオン]]タンパク質が示す伝播能力とも関連していると考えられている。すなわち、一旦異常構造をとったタンパク質がシードとなり、別の個体におけるアミロイドタンパク質の構造及び性質を変化させていくというモデルである<ref><pubmed> 8513491 </pubmed></ref>。またシードへの組み込みはアミロイドタンパク質が同様の構造を取りうるかどうかに依存する。プリオンの感染性にはごく僅かなアミノ酸の違いに起因する「種の壁」が存在するが、この現象も一次配列の違いに依存する各種のプリオンが形成するシード構造の違いによって説明できる。 | ||
| 165行目: | 169行目: | ||
このようなタンパク質凝集物の細胞間伝播という概念は必ずしもアミロイドの形成には依存しておらず、凝集して線維を形成するタンパク質に普遍的に観察される可能性があり、最近では様々な神経変性疾患において細胞内に蓄積するタンパク質([[タウ]]、[[シヌクレイン]]、[[TDP-43]]など)においても伝播能力の存在が確認されつつある<ref><pubmed> 24005412 </pubmed></ref>。また[[wikipedia:ja:酵母|酵母]]などにおいてはプリオン様タンパク性因子による形質転換が報告されており、タンパク質の構造変化に依存した形質の伝播様式として注目されている<ref><pubmed> 23379365 </pubmed></ref>。 | このようなタンパク質凝集物の細胞間伝播という概念は必ずしもアミロイドの形成には依存しておらず、凝集して線維を形成するタンパク質に普遍的に観察される可能性があり、最近では様々な神経変性疾患において細胞内に蓄積するタンパク質([[タウ]]、[[シヌクレイン]]、[[TDP-43]]など)においても伝播能力の存在が確認されつつある<ref><pubmed> 24005412 </pubmed></ref>。また[[wikipedia:ja:酵母|酵母]]などにおいてはプリオン様タンパク性因子による形質転換が報告されており、タンパク質の構造変化に依存した形質の伝播様式として注目されている<ref><pubmed> 23379365 </pubmed></ref>。 | ||
一方、アルツハイマー病患者脳から得られたAβ線維の構造解析がなされ、<i>in vitro</i>で凝集させた構造とは異なる凝集形態を示していたことから、<i>in vivo</i>における凝集プロセスの違いが指摘されており<ref><pubmed> 24034249 </pubmed></ref>、伝播メカニズムとの関係の解明が待たれている。 | |||
===細胞毒性=== | ===細胞毒性=== | ||
アミロイド線維が発揮する細胞障害および毒性はアミロイドーシスにおける臓器不全の基本的病態と言える。アミロイド沈着後に生じる疾患プロセスを抑制する治療薬の開発のためにも、その理解は必須である。しかしアミロイドタンパク質のどのような構造、分子状態が毒性を発揮するのかについては未だ明確ではない。近年ではAβとFAD変異がもたらす分子病態の解析から、アミロイド線維そのものではなく、その中間体となるオリゴマー<ref><pubmed> 12702875 </pubmed></ref>に起因しているというオリゴマー仮説が提唱されている。 | アミロイド線維が発揮する細胞障害および毒性はアミロイドーシスにおける臓器不全の基本的病態と言える。アミロイド沈着後に生じる疾患プロセスを抑制する治療薬の開発のためにも、その理解は必須である。しかしアミロイドタンパク質のどのような構造、分子状態が毒性を発揮するのかについては未だ明確ではない。近年ではAβとFAD変異がもたらす分子病態の解析から、アミロイド線維そのものではなく、その中間体となるオリゴマー<ref><pubmed> 12702875 </pubmed></ref>に起因しているというオリゴマー仮説が提唱されている。 | ||
このアミロイドタンパク質の凝集物がどのように細胞傷害を惹起しているか、という点については、[[脂質二重膜]]の障害、[[酸化的ストレス]]や[[小胞体ストレス]]の惹起、[[ミトコンドリア]]障害などが想定されている<ref><pubmed> 23820032 </pubmed></ref>。興味深いことに、全く異なるアミロイド原性タンパク質であるAβとADanが脳実質に蓄積するそれぞれの疾患モデルマウスを、神経障害と関連するtauトランスジェニックマウスと交配すると、いずれの場合もtau病理が亢進されることが示された<ref><pubmed> 20385796 </pubmed></ref>。これは少なくとも[[大脳皮質]] | このアミロイドタンパク質の凝集物がどのように細胞傷害を惹起しているか、という点については、[[脂質二重膜]]の障害、[[酸化的ストレス]]や[[小胞体ストレス]]の惹起、[[ミトコンドリア]]障害などが想定されている<ref><pubmed> 23820032 </pubmed></ref>。興味深いことに、全く異なるアミロイド原性タンパク質であるAβとADanが脳実質に蓄積するそれぞれの疾患モデルマウスを、神経障害と関連するtauトランスジェニックマウスと交配すると、いずれの場合もtau病理が亢進されることが示された<ref><pubmed> 20385796 </pubmed></ref>。これは少なくとも[[大脳皮質]]に沈着するアミロイドが示す神経細胞傷害プロセスの下流には共通性があることを示唆している。すなわち、アミロイド原性タンパク質の種類を問わず、どのような線維がどの細胞や臓器に沈着するかによって最終的にアミロイドーシスにおける病態が決定する可能性が考えられている。またAβが細胞外から神経細胞毒性を呈するために毒性受容体が想定さられており、[[NMDA型グルタミン酸受容体|NMDA型]]および[[AMPA型グルタミン酸受容体]]、α7[[ニコチン性アセチルコリン受容体]]、[[インスリン受容体]]、[[RAGE]]、プリオンタンパク質や[[EPHB2|EphB2]]、[[LilrB2]]などがその候補として挙げられている。 | ||
==関連項目== | ==関連項目== | ||
2014年1月3日 (金) 21:59時点における版
英:amyloidosis
アミロイドamyloidはコンゴーレッド染色でオレンジ色に染まり、偏光顕微鏡で緑色偏光を呈し、電子顕微鏡観察下では7~15nmの繊維構造を呈する物質である。アミロイドが、組織間隙に沈着して臓器の機能不全が生じる疾患をアミロイドーシス amyloidosisと呼ぶ[1]。アミロイドタンパク質の種類や臓器によって特徴が見られ、大きく全身性アミロイドーシスと限局性アミロイドーシスに分類される。代表的な全身性アミロイドーシスには、全身性AAアミロイドーシス、家族性アミロイドニューロパチーが挙げられる。限局性アミロイドーシスには脳アミロイドーシスである、アルツハイマー病、脳血管アミロイドアンギオパチー、遺伝性アミロイド性脳出血で、クロイツフェルト・ヤコブ病などが知られている。基本的には、アミロイドーシス発症の分子病態は凝集するアミロイドタンパク質の濃度上昇か、凝集能亢進によるものである。したがってアミロイドタンパク質の除去が根本治療戦略となる。
アミロイドーシスとは
アミロイドamyloidはコンゴーレッド染色でオレンジ色に染まり、偏光顕微鏡で緑色偏光を呈し、電子顕微鏡観察下では7~15nmの繊維構造を呈する物質として定義される。アミロイドが、組織間隙に沈着する疾患を総称してアミロイドーシス amyloidosisと呼ぶ[2]。多くの場合、前駆タンパクであるアミロイドタンパク質が折りたたみ障害を引き起こして重合し、βシート構造に富む不溶性線維として蓄積・凝集している
沈着するアミロイドタンパク質の種類や臓器によって特徴が見られ、特に大きく全身性アミロイドーシスと限局性アミロイドーシスに分類されている。
全身性アミロイドーシス
アミロイドタンパク質が血中に存在する場合は全身性アミロイドーシスとなる[3]。
アミロイドタンパク質としては、モノクローナル免疫グロブリンのL鎖由来のアミロイドALやH鎖由来のアミロイドAH、血清アミロイドAの代謝産物であるアミロイドA(AA)、β2ミクログロブリン、トランスサイレチン、ゲルソリン、アポAIが知られている。いずれもアミロイドタンパク質の産生亢進、濃度上昇がアミロイドーシスを惹起していることが知られており、例えばアミロイドALでは免疫グロブリン産生細胞である形質細胞の過剰な増殖や腫瘍化がその原因である。また膠原病やリウマチなどが原因となり全身性慢性炎症を基礎疾患として血清アミロイドAの濃度上昇が継続し、全身性AAアミロイドーシスを惹起する。さらに腎障害及び血液透析によってβ2ミクログロブリンの排泄、除去が不全となり、10年以上の長期透析の結果アミロイド沈着を招くことが知られている。
遺伝子変異によって生じる全身性アミロイドーシスとして、家族性アミロイドニューロパチー Familial amyloid polyneuropathy(FAP)が知られている[4]。FAPはトランスサイレチン、ゲルソリン、アポAI、血清アミロイドA遺伝子変異に連鎖し、これらのアミロイドタンパク質が神経節を含む神経系および他の臓器に沈着する。また最近になり、全身性アミロイドーシスを惹起するプリオン遺伝子変異も同定された[5]。我が国を含めて、特にトランスサイレチン遺伝子変異によるFAPが最も多い[6]。
通常トランスサイレチンは四量体を形成しているが、遺伝子変異によって生じるアミノ酸置換によって不安定な単量体へ解離しやすくなり、なんらかの機序で重合して線維化すると考えられている。体内のトランスサイレチンは主として肝臓で産生されるが、肝実質にアミロイドは沈着しない。このためFAP患者の肝臓を移植により正常肝に換えることでアミロイドタンパク質である変異トランスサイレチンの消失が期待され、移植後多くの症例でFAPの臨床進行が停止するか、遅延することが確認されている。また2013年には、トランスサイレチンの四量体の解離及び変性を抑制することでアミロイド形成を阻害し、末梢神経障害の進行を抑制するVyndaqel(一般名:Tafamidis)が承認された。
トランスサイレチンが全身性に蓄積する疾患として、老人性全身性アミロイドーシスも知られている。この疾患では心室へ大量にアミロイド沈着を生じ難治性の不整脈と心不全をきたし、老年期心疾患の重要なものの一つである。高齢者の心臓へのアミロイド沈着は80歳以上の剖検例では25~28%の頻度でみられ、心房細動から最終的には心不全に陥る。確定診断としてはトランスサイレチン遺伝子に変異はなく、野生型トランスサイレチンの蓄積が見られることが挙げられる。
限局性アミロイドーシス
特定の臓器に限局して沈着を認める場合は限局性アミロイドーシスとなる。臓器に応じて分類され、脳アミロイドーシス[7]としてはアルツハイマー病や脳血管アミロイドアンギオパチーで蓄積が見られるアミロイドβタンパク質(Aβ)の他、シスタチンCの遺伝子変異[8]がアイスランド型遺伝性アミロイド性脳出血で見出されている。
またプリオンタンパク質の蓄積、沈着はクロイツフェルト・ヤコブ病やゲルストマン・ストロイスラー・シャインカー症候群などのプリオン病患者脳で報告されている。さらにBRI2遺伝子の変異によって生じるアミロイドペプチドABri、ADanはそれぞれBritish型、Danish型家族性認知症患者脳において蓄積している[9]。BRI2はその最C末端部がFurinによって切断され分泌されているが、野生型ペプチドには凝集性が認められない。しかし終止コドン近傍の遺伝子変異により野生型よりも僅かに長く、凝集性の高いペプチドが分泌され、これらがアミロイドとして脳実質に蓄積する。
その他の限局性アミロイドーシスとしては、内分泌アミロイドーシスのアミロイドタンパク質としてはカルシトニン、アミリン、インスリン、心房ナトリウム利尿ペプチドが同定されており、主にこれらのホルモンを分泌する細胞由来の腫瘍内で蓄積・沈着が観察される。また皮膚アミロイドーシスとしてはケラチンが、限局性結節性アミロイドーシスはアミロイドALがアミロイドタンパク質として蓄積することが報告されている。
| タイプ | 頻度 | 詳細 |
|---|---|---|
| 原発性アミロイドーシス | 稀 | 基礎疾患を伴わないアミロイドーシス |
| 家族性アミロイドーシス | 稀 | 遺伝的に変異をおこしたアミロイド原性タンパク質によって惹起されるアミロイドーシス |
| 二次性アミロイドーシス | 低 | 何らかの基礎疾患に合併して生じるアミロイドーシス 多発性骨髄腫や原発性マクログロブリン血症とALアミロイドーシス 関節リウマチとAAアミロイドーシス 長期透析とAβ2Mアミロイドーシス |
| 老人性全身アミロイドーシス | 高 | 加齢とともに心臓を含めた全身においてトランスサイレチンが蓄積する 遺伝子変異を伴わず、野生型アミロイドタンパク質が蓄積する |
| 略称 | アミロイドタンパク質 | 病態 | OMIM |
|---|---|---|---|
| AL | 免疫グロブリンL鎖 | 異常形質細胞によって産出されるモノクローナル免疫グロブリンL鎖由来のアミロイドALが全身諸臓器(心臓、腎臓、消化管、肝臓、末梢神経など)に沈着する。 | 254500 |
| AA | 血清アミロイドA | 慢性炎症時におもに肝臓から産出される急性期タンパク質の血清アミロイドA(SAA)の代謝産物アミロイドA(AA)が腎臓や消化管に沈着する。 | |
| Aβ | アミロイドβタンパク質 | アルツハイマー病患者脳において蓄積する老人斑はAβを主要構成成分とする。健常高齢者脳においても蓄積が見られることがある。 | 605714 |
| ATTR | トランスサイレチン | 主として肝臓から産生されるが、トランスサイレチン遺伝子に変異のある異型TTRは肝実質にアミロイド沈着はほとんどなく、神経節を含む末梢神経、自律神経系やほかの組織に沈着する。 野生型トランスサイレチンが蓄積する老人性全身性アミロイドーシスでは主に心臓、他に肺、腎臓、全身の小血管にアミロイドが蓄積する。 |
105210 |
| Aβ2M | β2ミクログロブリン | 長期透析患者では血中で増加しているβ2ミクログロブリン由来のアミロイドが靱帯、骨領域に沈着する。 | |
| AIAPP | アミリン | Ⅱ型糖尿病に伴い膵ランゲルハンス島やインスリノーマにアミリン由来のアミロイドが蓄積する。 | |
| APrP | プリオン | プリオン病においては異常なフォールディングを受けたプリオンタンパク質が脳内に蓄積する。 | 123400 |
| AGel | ゲルソリン | ゲルソリン遺伝子変異により、アミロイド蓄積が惹起される。 | 105120 |
| ACys | シスタチンC | シスタチンC遺伝子変異により、脳血管周囲にアミロイドが蓄積するアミロイドアンギオパチーを家族性に発症する。 | 105150 |
| AApoA1 | アポAI | ApoA1遺伝子変異により、主に腎臓にアミロイドが蓄積するFamilial visceral amyloidosisを発症する。 | 105200 |
| AFib | フィブリノーゲンα鎖 | FGA遺伝子変異により、主に腎臓にアミロイドが蓄積するFamilial visceral amyloidosisを発症する。 | 105200 |
| ALys | リゾチーム | LYZ遺伝子変異により、主に腎臓にアミロイドが蓄積するFamilial visceral amyloidosisを発症する。 | 105200 |
| ? | オンコスタチンM受容体 | OSMR遺伝子変異により皮膚に生じるアミロイドーシス。 | 105250 |
| ABri ADan |
BRI2/ITM2B | BRI2/ITM2B遺伝子変異により生じる脳アミロイドーシス。 | 176500 117300 |
| APro | プロラクチン | 下垂体のプロラクチン産生腺腫において見出されるアミロイドーシス。 | |
| AKer | ケラチン(ケラトエピセリン) | 皮膚に限局して発症するアミロイドーシス。 | |
| AANF | 心房性ナトリウム利尿ペプチド | 心房に限局して沈着するアミロイドーシス。 | |
| ACal | カルシトニン | 甲状腺髄様癌において見出されるアミロイドーシス。 |
病態生理
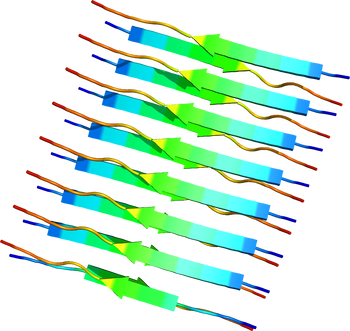
トランスサイレチン部分ペプチドからなるクロスβ構造。PDB ID: 2M5N
(編集コメント:結晶構造は回転できる物に取り替えました。これで良いかご確認下さい。SafariではWebGLをonにして下さい。)
構造
各アミロイドタンパク質には一定の共通したアミノ酸配列や構造は見られないが、アミロイド線維になると共通してクロスβ構造と呼ばれる形態をとっている[10][11][12]。これはアミロイド線維を構成するポリペプチド鎖が線維軸と垂直方向にβストランドとなり、かつ線維軸方向にβシート構造をとっているものである。このような構造学的特徴はイメージング技術に応用されつつあり、Aβ線維に特異的に結合する低分子化合物を利用したアミロイドPETスキャンが可能となった[13][14]。
線維形成過程と伝播

アミロイド線維形成過程におけるシードの役割

患者脳由来アミロイドから伸長したアミロイドβタンパク質の分子構造。PDB ID: 2M4J
(編集コメント:長かったので、小見出しをつけました。内容に照らして適当かご確認下さい。)
アミロイド線維形成過程では、多くの場合正常なフォールディングをうけているアミロイドタンパク質が何らかの理由で一旦部分変性し、会合することが必要である。また線維形成過程はその鋳型となるシード(種、核)の形成を契機として急速に進んでいくことが示されている[15]。すなわち、このシードの両端の末端にアミロイドタンパク質が結合して線維が伸長していくと考えられている。
このようなシード依存性伸長反応モデルは、プリオンタンパク質が示す伝播能力とも関連していると考えられている。すなわち、一旦異常構造をとったタンパク質がシードとなり、別の個体におけるアミロイドタンパク質の構造及び性質を変化させていくというモデルである[16]。またシードへの組み込みはアミロイドタンパク質が同様の構造を取りうるかどうかに依存する。プリオンの感染性にはごく僅かなアミノ酸の違いに起因する「種の壁」が存在するが、この現象も一次配列の違いに依存する各種のプリオンが形成するシード構造の違いによって説明できる。
最近ではアミロイドを形成しうるアミロイドタンパク質がいずれもプリオン様の伝播能力を示す可能性が推測されている[17][18]。実際、全身性アミロイドーシスの一つであるAAアミロイドーシスはモデルマウスを用いた伝播実験が確認されているが、野生のチーターにおいてAAアミロイドーシス発症頻度が近年上昇していることが示されていた。そして興味深いことに、AAアミロイドーシスを発症した個体の糞に伝播性が極めて高いアミロイドA線維が含まれていることが明らかとなった[19]。
糞便を介したアミロイドーシス伝播は、野生動物におけるプリオン病(ヒツジおけるスクレイピー、シカにおけるChronic wasting disease)の水平伝播メカニズムを説明できるものとして注目を集めている。特に末梢神経やリンパ節を介したプリオンの伝播に関しては、食物摂取などを介した末梢組織から生じうる限局性アミロイドーシスの発症機構を担っている可能性がある[20]。またAβについても、アルツハイマー病モデルマウスの腹腔内にAβ線維を注入すると大脳皮質でのAβの沈着が亢進することも示されている[21]。
このようなタンパク質凝集物の細胞間伝播という概念は必ずしもアミロイドの形成には依存しておらず、凝集して線維を形成するタンパク質に普遍的に観察される可能性があり、最近では様々な神経変性疾患において細胞内に蓄積するタンパク質(タウ、シヌクレイン、TDP-43など)においても伝播能力の存在が確認されつつある[22]。また酵母などにおいてはプリオン様タンパク性因子による形質転換が報告されており、タンパク質の構造変化に依存した形質の伝播様式として注目されている[23]。
一方、アルツハイマー病患者脳から得られたAβ線維の構造解析がなされ、in vitroで凝集させた構造とは異なる凝集形態を示していたことから、in vivoにおける凝集プロセスの違いが指摘されており[24]、伝播メカニズムとの関係の解明が待たれている。
細胞毒性
アミロイド線維が発揮する細胞障害および毒性はアミロイドーシスにおける臓器不全の基本的病態と言える。アミロイド沈着後に生じる疾患プロセスを抑制する治療薬の開発のためにも、その理解は必須である。しかしアミロイドタンパク質のどのような構造、分子状態が毒性を発揮するのかについては未だ明確ではない。近年ではAβとFAD変異がもたらす分子病態の解析から、アミロイド線維そのものではなく、その中間体となるオリゴマー[25]に起因しているというオリゴマー仮説が提唱されている。
このアミロイドタンパク質の凝集物がどのように細胞傷害を惹起しているか、という点については、脂質二重膜の障害、酸化的ストレスや小胞体ストレスの惹起、ミトコンドリア障害などが想定されている[26]。興味深いことに、全く異なるアミロイド原性タンパク質であるAβとADanが脳実質に蓄積するそれぞれの疾患モデルマウスを、神経障害と関連するtauトランスジェニックマウスと交配すると、いずれの場合もtau病理が亢進されることが示された[27]。これは少なくとも大脳皮質に沈着するアミロイドが示す神経細胞傷害プロセスの下流には共通性があることを示唆している。すなわち、アミロイド原性タンパク質の種類を問わず、どのような線維がどの細胞や臓器に沈着するかによって最終的にアミロイドーシスにおける病態が決定する可能性が考えられている。またAβが細胞外から神経細胞毒性を呈するために毒性受容体が想定さられており、NMDA型およびAMPA型グルタミン酸受容体、α7ニコチン性アセチルコリン受容体、インスリン受容体、RAGE、プリオンタンパク質やEphB2、LilrB2などがその候補として挙げられている。
関連項目
参考文献
- ↑
Radford, S.E., & Weissman, J.S. (2012).
Special issue: the molecular and cellular mechanisms of amyloidosis. Journal of molecular biology, 421(2-3), 139-41. [PubMed:22664198] [WorldCat] [DOI] - ↑
Radford, S.E., & Weissman, J.S. (2012).
Special issue: the molecular and cellular mechanisms of amyloidosis. Journal of molecular biology, 421(2-3), 139-41. [PubMed:22664198] [WorldCat] [DOI] - ↑
Blancas-Mejía, L.M., & Ramirez-Alvarado, M. (2013).
Systemic amyloidoses. Annual review of biochemistry, 82, 745-74. [PubMed:23451869] [PMC] [WorldCat] [DOI] - ↑
Planté-Bordeneuve, V., & Said, G. (2011).
Familial amyloid polyneuropathy. The Lancet. Neurology, 10(12), 1086-97. [PubMed:22094129] [WorldCat] [DOI] - ↑
Mead, S., Gandhi, S., Beck, J., Caine, D., Gallujipali, D., Carswell, C., ..., & Collinge, J. (2013).
A novel prion disease associated with diarrhea and autonomic neuropathy. The New England journal of medicine, 369(20), 1904-14. [PubMed:24224623] [PMC] [WorldCat] [DOI] - ↑
Ikeda, S., Nakazato, M., Ando, Y., & Sobue, G. (2002).
Familial transthyretin-type amyloid polyneuropathy in Japan: clinical and genetic heterogeneity. Neurology, 58(7), 1001-7. [PubMed:11940682] [WorldCat] [DOI] - ↑
Yamada, M., & Naiki, H. (2012).
Cerebral amyloid angiopathy. Progress in molecular biology and translational science, 107, 41-78. [PubMed:22482447] [WorldCat] [DOI] - ↑
Palsdottir, A., Abrahamson, M., Thorsteinsson, L., Arnason, A., Olafsson, I., Grubb, A., & Jensson, O. (1988).
Mutation in cystatin C gene causes hereditary brain haemorrhage. Lancet (London, England), 2(8611), 603-4. [PubMed:2900981] [WorldCat] [DOI] - ↑
Tsachaki, M., Ghiso, J., & Efthimiopoulos, S. (2008).
BRI2 as a central protein involved in neurodegeneration. Biotechnology journal, 3(12), 1548-54. [PubMed:19072909] [WorldCat] [DOI] - ↑
Sawaya, M.R., Sambashivan, S., Nelson, R., Ivanova, M.I., Sievers, S.A., Apostol, M.I., ..., & Eisenberg, D. (2007).
Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature, 447(7143), 453-7. [PubMed:17468747] [WorldCat] [DOI] - ↑
Toyama, B.H., & Weissman, J.S. (2011).
Amyloid structure: conformational diversity and consequences. Annual review of biochemistry, 80, 557-85. [PubMed:21456964] [PMC] [WorldCat] [DOI] - ↑
Fitzpatrick, A.W., Debelouchina, G.T., Bayro, M.J., Clare, D.K., Caporini, M.A., Bajaj, V.S., ..., & Dobson, C.M. (2013).
Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proceedings of the National Academy of Sciences of the United States of America, 110(14), 5468-73. [PubMed:23513222] [PMC] [WorldCat] [DOI] - ↑
Klunk, W.E., Engler, H., Nordberg, A., Wang, Y., Blomqvist, G., Holt, D.P., ..., & Långström, B. (2004).
Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Annals of neurology, 55(3), 306-19. [PubMed:14991808] [WorldCat] [DOI] - ↑
Clark, C.M., Schneider, J.A., Bedell, B.J., Beach, T.G., Bilker, W.B., Mintun, M.A., ..., & AV45-A07 Study Group (2011).
Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA, 305(3), 275-83. [PubMed:21245183] [WorldCat] [DOI] - ↑
Lee, Y.H., & Goto, Y. (2012).
Kinetic intermediates of amyloid fibrillation studied by hydrogen exchange methods with nuclear magnetic resonance. Biochimica et biophysica acta, 1824(12), 1307-23. [PubMed:22885025] [WorldCat] [DOI] - ↑
Jarrett, J.T., & Lansbury, P.T. (1993).
Seeding "one-dimensional crystallization" of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell, 73(6), 1055-8. [PubMed:8513491] [WorldCat] [DOI] - ↑
Nussbaum, J.M., Schilling, S., Cynis, H., Silva, A., Swanson, E., Wangsanut, T., ..., & Bloom, G.S. (2012).
Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature, 485(7400), 651-5. [PubMed:22660329] [PMC] [WorldCat] [DOI] - ↑
Jucker, M., & Walker, L.C. (2013).
Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature, 501(7465), 45-51. [PubMed:24005412] [PMC] [WorldCat] [DOI] - ↑
Zhang, B., Une, Y., Fu, X., Yan, J., Ge, F., Yao, J., ..., & Higuchi, K. (2008).
Fecal transmission of AA amyloidosis in the cheetah contributes to high incidence of disease. Proceedings of the National Academy of Sciences of the United States of America, 105(20), 7263-8. [PubMed:18474855] [PMC] [WorldCat] [DOI] - ↑
Aguzzi, A., Nuvolone, M., & Zhu, C. (2013).
The immunobiology of prion diseases. Nature reviews. Immunology, 13(12), 888-902. [PubMed:24189576] [WorldCat] [DOI] - ↑
Eisele, Y.S., Obermüller, U., Heilbronner, G., Baumann, F., Kaeser, S.A., Wolburg, H., ..., & Jucker, M. (2010).
Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science (New York, N.Y.), 330(6006), 980-2. [PubMed:20966215] [PMC] [WorldCat] [DOI] - ↑
Jucker, M., & Walker, L.C. (2013).
Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature, 501(7465), 45-51. [PubMed:24005412] [PMC] [WorldCat] [DOI] - ↑
Wickner, R.B., Edskes, H.K., Bateman, D.A., Kelly, A.C., Gorkovskiy, A., Dayani, Y., & Zhou, A. (2013).
Amyloids and yeast prion biology. Biochemistry, 52(9), 1514-27. [PubMed:23379365] [WorldCat] [DOI] - ↑
Lu, J.X., Qiang, W., Yau, W.M., Schwieters, C.D., Meredith, S.C., & Tycko, R. (2013).
Molecular structure of β-amyloid fibrils in Alzheimer's disease brain tissue. Cell, 154(6), 1257-68. [PubMed:24034249] [PMC] [WorldCat] [DOI] - ↑
Kayed, R., Head, E., Thompson, J.L., McIntire, T.M., Milton, S.C., Cotman, C.W., & Glabe, C.G. (2003).
Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science (New York, N.Y.), 300(5618), 486-9. [PubMed:12702875] [WorldCat] [DOI] - ↑
Cheng, B., Gong, H., Xiao, H., Petersen, R.B., Zheng, L., & Huang, K. (2013).
Inhibiting toxic aggregation of amyloidogenic proteins: a therapeutic strategy for protein misfolding diseases. Biochimica et biophysica acta, 1830(10), 4860-71. [PubMed:23820032] [WorldCat] [DOI] - ↑
Coomaraswamy, J., Kilger, E., Wölfing, H., Schäfer, C., Kaeser, S.A., Wegenast-Braun, B.M., ..., & Jucker, M. (2010).
Modeling familial Danish dementia in mice supports the concept of the amyloid hypothesis of Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America, 107(17), 7969-74. [PubMed:20385796] [PMC] [WorldCat] [DOI]