「グルタミン酸トランスポーター」の版間の差分
細 →サブファミリー |
細 →発現 |
||
| (2人の利用者による、間の16版が非表示) | |||
| 2行目: | 2行目: | ||
<font size="+1">[http://researchmap.jp/read0191613 田中 光一]</font><br> | <font size="+1">[http://researchmap.jp/read0191613 田中 光一]</font><br> | ||
''東京医科歯科大学''<br> | ''東京医科歯科大学''<br> | ||
DOI:<selfdoi /> | DOI:<selfdoi /> 原稿受付日:2016年1月26日 原稿完成日:2016年2月12日<br> | ||
担当編集委員:<br> | 担当編集委員:[http://researchmap.jp/wadancnp 和田 圭司](国立研究開発法人国立精神・神経医療研究センター)<br> | ||
</div> | </div> | ||
{{box|text= | {{box|text= グルタミン酸は哺乳類の中枢神経系において記憶・学習などの高次機能を調節する主要な興奮性神経伝達物質として知られている。一方、過剰なグルタミン酸は、グルタミン酸受容体の過剰な活性化によりグルタミン酸興奮毒性と呼ばれる神経細胞障害作用を持つことが知られている。このため細胞外グルタミン酸濃度は厳密に制御される必要があり、グルタミン酸トランスポーターがその役割を担う。これまで哺乳類の中枢神経系において、5種類のグルタミン酸トランスポーターサブファミリー、slc1a1、slc1a2、slc1a3、slc1a6、slc1a7が単離されている。slc1a2、slc1a3は主にアストロサイトに、slc1a1とslc1a6は神経細胞に、slc1a7は網膜に発現している。シナプス間隙におけるグルタミン酸の除去は、主にアストロサイトに存在する2種類のグルタミン酸輸送体slc1a2、slc1a3により担われている。近年、これらのグルタミン酸トランスポーターの機能障害が様々な精神神経疾患の発症に関与することが明らかになりつつある。}} | ||
{{Infobox protein family | |||
| Symbol = | |||
| Name =グルタミン酸トランスポーター | |||
| image = 1xfh.pdb | |||
| width = 250px | |||
| caption = グルタミン酸トランスポーターホモログ(''Pyrococcus horikoshii'') | |||
| Pfam = PF00375 | |||
| Pfam_clan = | |||
| InterPro = IPR001991 | |||
| SMART = | |||
| PROSITE = PDOC00591 | |||
| MEROPS = | |||
| SCOP = 1XFH | |||
| TCDB = 2.A.23.1.5 | |||
| OPM family = | |||
| OPM protein = 2nwl | |||
}} | }} | ||
==グルタミン酸トランスポーターとは== | ==グルタミン酸トランスポーターとは== | ||
| 16行目: | 30行目: | ||
[[image:グルタミン酸トランスポーター2.png|thumb|300px|'''図2.グルタミン酸トランスポーターの基本的構造''']] | [[image:グルタミン酸トランスポーター2.png|thumb|300px|'''図2.グルタミン酸トランスポーターの基本的構造''']] | ||
[[グルタミン酸]] | [[グルタミン酸]]は、哺乳類中枢神経系において約70%の神経細胞が用いる主要な興奮性神経伝達物質であり、[[記憶]]・[[学習]]などの脳高次機能に重要な役割を果たしている<ref name=ref1><pubmed>9651535</pubmed></ref>。しかし、その機能的な重要性の反面、[[興奮毒性]]という概念で表されるように<ref name=ref2><pubmed>2908446</pubmed></ref>、過剰なグルタミン酸は神経細胞障害作用を持ち、様々な[[精神神経疾患]]の発症に関与することが明らかになりつつある<ref name=ref3><pubmed>26569330</pubmed></ref> <ref name=ref4><pubmed>25482181</pubmed></ref>。従って、[[シナプス間隙]]におけるグルタミン酸濃度は厳密に制御されなければならない。 | ||
シナプスにおけるグルタミン酸の動態は図1のように考えられている。シナプス前終末から放出されたグルタミン酸は、シナプス後細胞のグルタミン酸受容体に結合しその効果を発揮するが、伝達終了後シナプス間隙のグルタミン酸は[[アストロサイト]]およびシナプス後神経細胞膜に存在するグルタミン酸トランスポーターにより細胞内に取り込まれる。アストロサイトに取り込まれたグルタミン酸は、[[グルタミン合成酵素]]によりグルタミンに変換され、グリア細胞外に放出され、[[グルタミン-グルタミン酸サイクル]]を経て、再びシナプス小胞に蓄えられる。 | |||
中枢神経系には5種類のグルタミン酸トランスポーターサブファミリーが存在することが知られおり<ref name=ref5><pubmed>11369436</pubmed></ref>、グルタミン酸トランスポーター欠損マウスの解析を通じ、グルタミン酸トランスポーターの各サブファミリーの機能的役割が明らかになりつつある。 | |||
==構造== | ==構造== | ||
5種類のグルタミン酸トランスポーターは互いに50~55%の相同性を有するアミノ酸数520~ | 5種類のグルタミン酸トランスポーターは互いに50~55%の相同性を有するアミノ酸数520~580のタンパク質である。グルタミン酸トランスポーターの構造はN末端とC末端は細胞内に位置し、8個の膜貫通部位、2個の膜を完全に貫通していないループ構造(re-entrant loop)を持つ(図2)<ref name=ref6><pubmed>20708631</pubmed></ref>。グルタミン酸トランスポーターはグルタミン酸を取り込む際に、Na<sup>+</sup>およびH<sup>+</sup>の[[共輸送]]、K<sup>+</sup>の[[対向輸送]]と共役する。グルタミン酸、Na<sup>+</sup>、H<sup>+</sup>、K<sup>+</sup>の結合・輸送に関与しているアミノ酸残基はいずれも6番目の膜貫通領域以降のC末端側である。また、グルタミン酸トランスポーターは3量体のホモオリゴマーとして細胞膜に発現している。 | ||
==サブファミリー== | ==サブファミリー== | ||
| 28行目: | 46行目: | ||
|- | |- | ||
|style="background-color:#f0fff0"| | |style="background-color:#f0fff0"| | ||
|style="background-color:#f0fff0"| | |style="background-color:#f0fff0"|別の名称 | ||
|style="background-color:#f0fff0"| | |style="background-color:#f0fff0"|アミノ酸残基数(ヒト) | ||
|style="background-color:#f0fff0"| | |style="background-color:#f0fff0"|組織分布 | ||
|style="background-color:#f0fff0"| | |style="background-color:#f0fff0"|中枢神経系での細胞局在 | ||
|style="background-color:#f0fff0"| | |style="background-color:#f0fff0"|遺伝子座(ヒト) | ||
|- | |- | ||
|style="background-color:#f0fff0"| | |style="background-color:#f0fff0"|slc1a1 | ||
|EAAT3<br>EAAC1 | |EAAT3<br>EAAC1 | ||
|524 | |||
|脳・心臓・腎臓・肝臓・小腸 | |||
|神経細胞 | |||
|9p24 | |||
|-| | |||
|style="background-color:#f0fff0"|slc1a2 | |||
|EAAT2<br>GLT-1 | |EAAT2<br>GLT-1 | ||
|574 | |||
|脳・肝臓・膵臓 | |||
|アストロサイト | |||
|11p13-p12 | |||
|-| | |||
|style="background-color:#f0fff0"|slc1a3 | |||
|EAAT1<br>GLAST | |EAAT1<br>GLAST | ||
|542 | |542 | ||
|脳・心臓・肺・骨格筋・胎盤 | |脳・心臓・肺・骨格筋・胎盤 | ||
|アストロサイト | |||
|5p13 | |||
|- | |||
|style="background-color:#f0fff0"|slc1a6 | |||
|EAAT4 | |||
|564 | |||
|脳・胎盤 | |脳・胎盤 | ||
|プルキンエ細胞 | |プルキンエ細胞 | ||
| | |19p13.12 | ||
|- | |- | ||
|style="background-color:#f0fff0"| | |style="background-color:#f0fff0"|slc1a7 | ||
| | |EAAT5 | ||
| | |560 | ||
| | |網膜・肝臓・心臓・肺・骨格筋・小腸 | ||
| | |視細胞・双極細胞 | ||
|1p32.3 | |1p32.3 | ||
|- | |- | ||
| 74行目: | 92行目: | ||
[[image:グルタミン酸トランスポーター3.png|thumb|300px|'''図3.グルタミン酸トランスポーターサブファミリーの脳内分布''']] | [[image:グルタミン酸トランスポーター3.png|thumb|300px|'''図3.グルタミン酸トランスポーターサブファミリーの脳内分布''']] | ||
5種類のグルタミン酸トランスポーターサブファミリーは、中枢神経系だけでなく末梢組織にも発現していることが知られているが、本稿では中枢神経系における発現に関して記述する(表1)。 | |||
[[slc1a2]]([[GLT-1]]/[[EAAT2]])は[[大脳皮質]]・[[海馬]]の[[アストロサイト]]に、[[slc1a3]]([[GLAST]]/[[EAAT1]])は[[小脳]]のアストロサイトに優位に発現している<ref name=ref7><pubmed>8733726</pubmed></ref>。[[slc1a1]]([[EAAC1]]/[[EAAT3]])は神経細胞に存在し、中枢神経系に広く分布している<ref name=ref7 />。[[slc1a6]]([[EAAT4]])は小脳の[[プルキンエ細胞]]に<ref name=ref8><pubmed>8905715</pubmed></ref>、また[[slc1a7]]([[EAAT5]])は[[網膜]]の[[視細胞]]・[[双極細胞]]に特異的に発現している<ref name=ref9><pubmed>10696802</pubmed></ref>(図3)。神経細胞に発現しているslc1a1とslc1a4は、神経細胞の終末ではなく[[細胞体]]・[[樹状突起]]に主に局在している<ref name=ref10><pubmed>7917301</pubmed></ref> <ref name=ref11><pubmed>9261809</pubmed></ref>。アストロサイトに発現しているslc1a2とslc1a3は、シナプス周囲を覆っている突起に密度高く局在している<ref name=ref12><pubmed>7546749</pubmed></ref>。 | |||
最近、[[CDC42EP4]]/[[septin]]がslc1a3をシナプス周囲を覆う[[バーグマングリア]]の突起に局在させることが明らかになった<ref name=ref13><pubmed>26657011</pubmed></ref>。成人脳ではアストロサイトに局在するslc1a2は、胎児期から生後3日の発生初期には、一過性に神経細胞に発現する<ref name=ref14><pubmed>9671661</pubmed></ref>。 | |||
また、成人脳ではアストロサイトに局在するslc1a3は、発生初期には[[神経幹細胞]]に発現しており、神経幹細胞のマーカーとして用いられている<ref name=ref15><pubmed>9364068</pubmed></ref> <ref name=ref16><pubmed>26586824</pubmed></ref>。 | |||
==機能== | ==機能== | ||
| 80行目: | 104行目: | ||
===分子機能=== | ===分子機能=== | ||
グルタミン酸トランスポーターは、細胞膜を介したNa+ | グルタミン酸トランスポーターは、細胞膜を介したNa<sup>+</sup>の[[電気化学ポテンシャル]]を利用して、グルタミン酸を輸送する。1分子のグルタミン酸の取り込みは、3個のNa<sup>+</sup>および1個のH<sup>+</sup>の共輸送、1個のK<sup>+</sup>の対向輸送と共役する(図2)。従って、グルタミン酸トランスポーターは[[起電性]]であり、グルタミン酸の細胞内への取り込みにより[[内向き電流]]が生じる。また、これとは別に、熱力学的にグルタミン酸取り込みと連動していないCl<sup>-</sup>の流入があることが知られているが、Cl<sup>-</sup>の透過性の順番はslc1a6/7 > slc1a3 > slc1a1 > slc1a2である<ref name=ref17><pubmed>26303507</pubmed></ref>。 | ||
slc1a1は、グルタミン酸の他に電荷をもたない[[L-システイン]]を取り込み、[[グルタチオン]]合成に利用している<ref name=ref18><pubmed>25275463</pubmed></ref>。 | |||
===生理機能=== | ===生理機能=== | ||
====海馬のシナプス伝達における役割==== | ====海馬のシナプス伝達における役割==== | ||
海馬において主要なグルタミン酸トランスポーターはslc1a2である。slc1a2欠損マウスの海馬の[[シェーファー側枝]]・[[CA1]][[錐体細胞]]間シナプスを電気生理学的に調べたところ、海馬のCA1錐体細胞で記録されるシェーファー側枝による[[興奮性シナプス後電流]]([[Excitatory Postsynaptic Current]]:[[EPSC]])の[[AMPA型グルタミン酸受容体|AMPA(α-amino-3-hydroxy-5-methylisoxazole-4- propionic acid)型グルタミン酸受容体]]成分・[[NMDA型グルタミン酸受容体]]成分とも、その振幅・時間経過は野生型と違いはなかった<ref name=ref19><pubmed>9180080</pubmed></ref>。このことは、GLT1は海馬において、EPSCの振幅・時間経過の重要な決定因子ではないことを示している。海馬におけるEPSCの振幅・時間経過は、[[グルタミン酸受容体]]自体のキネテクスにより規定されていると考えられる。海馬のシナプスは、[[グリア]]細胞の突起によるシナプス部位の被覆が不完全で、主に拡散がシナプス間隙におけるグルタミン酸のクリアランスを規定していると考えられる。 | |||
海馬CA1[[網状分子層]]の[[oriens-lacunosum moleculare]](O-LM)[[介在神経]]に多く存在する[[代謝型グルタミン酸受容体]][[mGluR1]]は、グリア型グルタミン酸トランスポーターslc1a2およびslc1a3により、活性が制御されている。slc1a2とslc1a3を抑制すると、mGluR1依存性EPSCの振幅が増加し、介在神経の発火が増強され、結果としてCA1錐体細胞の抑制が増強されたる<ref name=ref20><pubmed>15140926</pubmed></ref>。 | |||
また、slc1a2欠損マウスでは、海馬CA1領域のNMDA型グルタミン酸受容体成分が増強され、[[長期増強]]の発現が障害されている<ref name=ref21><pubmed>11553304</pubmed></ref>。逆に、slc1a2の発現を増加すると、[[苔状線維]]—[[CA3]]錐体細胞間シナプスの[[長期抑圧]]の発現が障害される<ref name=ref22><pubmed>19651762</pubmed></ref>。 | |||
====小脳のシナプス伝達における役割==== | ====小脳のシナプス伝達における役割==== | ||
小脳ではプルキンエ細胞にグルタミン酸トランスポーターslc1a1、slc1a6が、プルキンエ細胞を取り囲むバーグマングリア細胞にはslc1a2, slc1a3が発現している。欠損マウスの解析から、[[平行線維]]・プルキンエ細胞間シナプス、[[登上線維]]・プルキンエ細胞間シナプスにおいて、グルタミン酸が放出された直後の高濃度のグルタミン酸の除去はslc1a3により、放出されてしばらく時間が経過した後の低濃度のグルタミン酸の除去はslc1a6により行われていることがわかった<ref name=ref23><pubmed>16177048</pubmed></ref> <ref name=ref24><pubmed>16377014</pubmed></ref>。さらにslc1a1とslc1a6の選択的機能阻害により隣接するシナプスへのグルタミン酸[[spillover]]が起こり、mGluR1依存性EPSCが増強、長期抑圧が促進される<ref name=ref25><pubmed>17727989</pubmed></ref> <ref name=ref26><pubmed>19583702</pubmed></ref>。また、slc1a6は、平行線維・バーグマングリア間のグルタミン酸伝達を制御している<ref name=ref27><pubmed>22302796</pubmed></ref>。 | |||
====大脳皮質のシナプス伝達の維持における役割==== | ====大脳皮質のシナプス伝達の維持における役割==== | ||
神経系は他の臓器に比べエネルギー要求性が高く、そのほとんどはシナプス伝達に使われる。従って、シナプス伝達を維持するためには、活動の亢進した部位に選択的にエネルギーを補充する必要がある。グリア型グルタミン酸トランスポーターslc1a2・slc1a3は、シナプス伝達のセンサーとして働き、神経活動の亢進→シナプス間隙のグルタミン酸濃度上昇→グリア型グルタミン酸トランスポーターによるグルタミン酸の再吸収(同時にNa<sup>+</sup>がグリア内へ流入)→グリアの[[Na+-K+ ATPase|Na<sup>+</sup>-K<sup>+</sup> ATPase]]の活性化(グリア内でのエネルギー消費増大)→グリアによる毛細血管からの[[ブドウ糖]]の取り込み増加→グリアの解糖系によるブドウ糖から乳酸の生成(グリア内の消費したエネルギーの補充)→生成した乳酸を神経細胞が取り込みエネルギーを補充、という一連のエネルギー補給反応をトリガーする(図4)<ref name=ref28><pubmed>12546822</pubmed></ref> <ref name=ref29><pubmed>16197522</pubmed></ref>。 | |||
====リボンシナプスにおける役割==== | ====リボンシナプスにおける役割==== | ||
[[リボンシナプス]]では、[[シナプス前細胞]]は[[活動電位]]を出さず、代わりに[[膜電位]]を連続的に変化させることで[[伝達物質]]の放出量を変化させ、情報を伝達する。リボンシナプスは網膜や[[内耳]]などの一次[[知覚]]のシナプスに存在する。 | |||
聴覚の一次知覚シナプスである[[内有毛細胞]]—[[蝸牛神経]]間シナプスは典型的なリボンシナプスであり、グルタミン酸が神経伝達物質である。内有毛細胞周囲の[[支持細胞]](inner phalangeal cell, IPC)にはslc1a3が、 [[蝸牛神経節]]細胞にはslc1a1とslc1a2が存在する。欠損マウスや薬理学的解析の結果から、内有毛細胞—蝸牛神経間シナプスのグルタミン酸の除去はIPCに存在するslc1a3により行われていることが明らかになった<ref name=ref30><pubmed>11102482</pubmed></ref> <ref name=ref31><pubmed>16855093</pubmed></ref> | |||
また、典型的なリボンシナプスの一つである網膜の[[視細胞]]—[[双極細胞]]間シナプスでは、視細胞に存在するグルタミン酸トランスポーターslc1a7がシナプス間隙からのグルタミン酸除去に主要な役割を果たす<ref name=ref32><pubmed>16600856</pubmed></ref>。 | |||
====脳形成における役割==== | ====脳形成における役割==== | ||
胎児期の脳には、slc1a1、slc1a2、slc1a3の3種類のグルタミン酸トランスポーターが存在する。これら3種類のサブファミリーの単独欠損マウスおよびslc1a1とslc1a3、slc1a1とslc1a2ダブル欠損マウスは正常な脳形成を示すが、slc1a2とslc1a3ダブル欠損マウスは胎生17日頃に死亡し、海馬・大脳皮質に層形成異常や大脳皮質と他の脳部位を結合する線維連絡に異常が観察される<ref name=ref33><pubmed>16880397</pubmed></ref>。これらの異常はNMDA型グルタミン酸受容体サブユニット1の欠損により改善する<ref name=ref34><pubmed>22606296</pubmed></ref>。細胞外に過剰に存在するグルタミン酸は、NMDA型グルタミン酸受容体を過剰に活性化し、神経細胞の移動、神経幹細胞の分裂、神経突起の伸長を障害する。従って、脳が正常に発達するには、slc1a2とslc1a3による細胞外グルタミン酸濃度の厳密な制御が必要である。 | |||
==疾患との関わり== | ==疾患との関わり== | ||
[[image:グルタミン酸トランスポーター5.png|thumb|300px|'''図5.グルタミン酸トランスポーターの機能異常による興奮と抑制のアンバランスが様々な精神神経疾患を引き起こす''']] | [[image:グルタミン酸トランスポーター5.png|thumb|300px|'''図5.グルタミン酸トランスポーターの機能異常による興奮と抑制のアンバランスが様々な精神神経疾患を引き起こす''']] | ||
脳の興奮性亢進は、多くの精神神経疾患に共通する病態と考えられている(図5)。脳の興奮性を亢進させる原因として、グルタミン酸シナプス伝達の亢進とGABAシナプス伝達の低下がある。グルタミン酸トランスポーターの機能障害は、グルタミン酸シナプス伝達を亢進させる主要な原因の一つである。 | |||
===統合失調症=== | ===統合失調症=== | ||
[[統合失調症]]は、[[幻聴]]・[[妄想]]などの[[陽性症状]]と、[[感情鈍麻]]、[[意欲]]の減退などの[[陰性症状]]、[[作業記憶]]などの[[認知障害]]を示し、有病率約1%の[[精神疾患]]である。統合失調症患者の遺伝子解析から、slc1a3遺伝子座の欠失やslc1a2の機能障害を伴うミスセンス変異を持つ症例が報告された<ref name=ref35><pubmed>18369103</pubmed></ref> <ref name=ref36><pubmed>22863191</pubmed></ref>。また、死後脳の解析からも、slc1a2およびslc1a3の発現が減少することが報告されている<ref name=ref37><pubmed>23356950</pubmed></ref>。slc1a3欠損マウスは、統合失調症の陽性症状・陰性症状・認知障害に相当する行動異常を示し、slc1a3の異常による脳の興奮性亢進が統合失調症の発症に重要な役割を果たすと考えられる<ref name=ref38><pubmed>18550032</pubmed></ref> <ref name=ref39><pubmed>19078949</pubmed></ref>。さらに、統合失調症の前駆期から初発期への移行に細胞外グルタミン酸濃度の上昇が関与することが報告されている<ref name=ref40><pubmed>24108440</pubmed></ref> <ref name=ref41><pubmed>23583108</pubmed></ref>。 | |||
===うつ病=== | ===うつ病=== | ||
[[うつ病]]は、抑うつ気分と興味・喜びの喪失などを特徴とする精神疾患である。近年、うつ病においても、以下のようなグルタミン酸神経伝達の亢進を示唆する知見が報告され、一つの仮説を形成しつつある<ref name=ref42><pubmed>18425072</pubmed></ref> <ref name=ref43><pubmed>21827775</pubmed></ref>。 | |||
# | #NMDA型グルタミン酸受容体阻害剤である[[ケタミン]]が、うつ病患者に即効性の抗うつ作用を示す<ref name=ref42 />。 | ||
#うつ病患者の血中・[[脳脊髄液]]中・脳内のグルタミン酸濃度は上昇している | #うつ病患者の血中・[[脳脊髄液]]中・脳内のグルタミン酸濃度は上昇している<ref name=ref44><pubmed>17574216</pubmed></ref> <ref name=ref45><pubmed>16707201</pubmed></ref>。 | ||
# | #うつ病患者の死後脳ではslc1a2とslc1a3の発現が減少している<ref name=ref46><pubmed>16230605</pubmed></ref>。 | ||
# | #slc1a2の発現を増加させる[[wikipedia:ja:|β-ラクタム系]][[wikipedia:ja:|抗生物質]]が、[[マウス]]のうつ様行動を改善する<ref name=ref47><pubmed>16860779</pubmed></ref>。 | ||
# | #slc1a2とslc1a3を活性化する[[リルゾール]]は、うつ病に効果がある<ref name=ref48><pubmed>17141740</pubmed></ref>。 | ||
手綱核特異的slc1a2欠損マウスは、うつ病の症状に似た行動異常や[[睡眠障害]]を起こす | 手綱核特異的slc1a2欠損マウスは、うつ病の症状に似た行動異常や[[睡眠障害]]を起こす<ref name=ref49><pubmed>25471567</pubmed></ref>。これらの知見は、グルタミン酸トランスポーターの機能不全によるグルタミン酸神経伝達の過剰な活性化が、うつ病の発症に重要な役割を果たしていることを示唆している。 | ||
===双極性障害=== | ===双極性障害=== | ||
[[双極性障害]] | [[双極性障害]]は、うつ状態と躁状態を繰り返す慢性の疾患である。[[1H-MRS|<sup>1</sup>H-MRS]]([[magnetic resonance spectroscopy]])によるグルタミン酸濃度測定のメタ解析で、双極性障害の患者において脳のグルタミン酸濃度が増加していることが報告されている<ref name=ref50><pubmed>22834460</pubmed></ref>。また、患者死後脳におけるslc1a2の発現減少や、双極性障害患者にのみ見られるslc1a2の稀なミスセンス変異(G6S、A20S、R31Q)が報告されている<ref name=ref51><pubmed>25406999</pubmed></ref>。さらに、NMDA型グルタミン酸受容体の阻害剤である[[メマンチン]]が躁病エピソードの治療に効果があるという報告もなされている<ref name=ref52><pubmed>25540723</pubmed></ref>。 | ||
===強迫性障害=== | ===強迫性障害=== | ||
[[強迫性障害]] | [[強迫性障害]]は、[[強迫観念]]・[[強迫行為]]を特徴とする疾患である。多くは思春期過ぎから発症し、人口の2~3%が罹患歴を持つ。近年、強迫性障害においても、以下のようなグルタミン酸神経伝達の亢進を示唆する知見が報告されている。 | ||
#強迫性障害患者の脳内ではグルタミン酸濃度が増加し、神経伝達が亢進している | #強迫性障害患者の脳内ではグルタミン酸濃度が増加し、神経伝達が亢進している<ref name=ref53><pubmed>19768139</pubmed></ref>。 | ||
# | #slc1a3の[[一塩基多型]]と強迫性障害の関連が報告されている<ref name=ref54><pubmed>24821223</pubmed></ref>。 | ||
#グルタミン酸神経伝達を抑制する薬剤に強迫性障害の治療効果がある | #グルタミン酸神経伝達を抑制する薬剤に強迫性障害の治療効果がある<ref name=ref53 />。 | ||
===自閉スペクトラム症=== | ===自閉スペクトラム症=== | ||
[[自閉スペクトラム症]] | [[自閉スペクトラム症]]は、[[社会性行動]]の喪失・[[言語発達]]の遅延・限局した興味や繰り返し行動を特徴とする脳高次機能の発達障害である。 | ||
自閉症様の行動を示す[[脆弱X症候群]]や[[結節性硬化症]]の患者ではグルタミン酸神経伝達の亢進が報告されている。自閉症のゲノムワイドな連鎖解析により、11番染色体の11p12-13が自閉症に関連があることが明らかになった<ref name=ref55><pubmed>17322880</pubmed></ref>。この領域はslc1a2の遺伝子座の近傍である。また、slc1a2を性成熟期以降に欠損させたマウスでは、大脳皮質—[[線条体]]間のグルタミン酸伝達が過剰に活性化され、過度な毛繕い行動と突発的に全身を激しく震わせるwet-dog shakeと呼ばれる繰り返し行動の回数が大幅に増加することが報告されている<ref name=ref56><pubmed>25662838</pubmed></ref>。 | |||
===てんかん=== | ===てんかん=== | ||
[[てんかん]]は、ニューロンの過剰な活動に伴い痙攣や意識障害などが発作的に反復して起こる慢性的な脳疾患である。てんかん発作の発現機序として、脳内の[[抑制性]]と興奮性神経伝達の不均衡状態が重要だと考えられている。[[内側側頭葉てんかん]]患者の海馬において、細胞外グルタミン酸濃度が上昇することが報告されている。しかし、[[側頭葉てんかん]]患者の外科切除海馬標本を用いた解析では、グルタミン酸トランスポーターslc1a2とslc1a3の発現減少に関して統一した結果が得られていない。グルタミン酸トランスポーターの遺伝子解析では、[[てんかん発作を伴うエピソード性運動失調6型]](Episodic ataxia with hemiplegic migraine and seizures)の患者にslc1a3の機能を障害するミスセンス変異(C186S とP290R)が見つかっている<ref name=ref57><pubmed>16116111</pubmed></ref> <ref name=ref58><pubmed>19139306</pubmed></ref>。 | |||
また、slc1a2欠損マウスは、致死性の自発てんかん発作により、生後3週齢から突然死を起こす。てんかん発作パターンは、[[NMDA]]を皮下注した時に見られる発作と類似しており、突然ケージの中を走り回り反弓緊張様姿勢をとり死亡する<ref name=ref19 />。さらに、slc1a3欠損マウスは、自発性てんかん発作は観察されないが、pentylentetorazole(PTZ)誘発性てんかんに対する感受性が亢進している<ref name=ref59><pubmed>10529447</pubmed></ref>。また、slic1a2の発現を増加させる薬物は、様々なてんかんモデルにおいて、抗てんかん作用を示すことが報告されている<ref name=ref60><pubmed>22513140</pubmed></ref> <ref name=ref61><pubmed>24569372</pubmed></ref>。 | |||
===薬物依存=== | ===薬物依存=== | ||
[[モルヒネ]]、[[コカイン]]、[[覚せい剤]] | [[モルヒネ]]、[[コカイン]]、[[覚せい剤]]、[[アルコール]]などの[[依存性薬物]]は、その反復摂取により依存性が形成される。近年の薬物乱用の低年齢化や一般市民への拡大は、社会的にも大きな問題となっている。[[薬物依存]]の形成・維持・再燃過程に、グルタミン酸神経伝達の亢進が重要な役割を果たすことは、以下の知見から示唆されている<ref name=ref62><pubmed>23876150</pubmed></ref> <ref name=ref63><pubmed>26033496</pubmed></ref> <ref name=ref64><pubmed>24741055</pubmed></ref>。 | ||
#薬物依存モデルの脳において細胞外グルタミン酸濃度が増加する。 | #薬物依存モデルの脳において細胞外グルタミン酸濃度が増加する。 | ||
#薬物依存モデルの脳においてslc1a2の発現量が減少する。 | #薬物依存モデルの脳においてslc1a2の発現量が減少する。 | ||
| 149行目: | 177行目: | ||
===アルツハイマー病=== | ===アルツハイマー病=== | ||
[[アルツハイマー病]] | [[アルツハイマー病]]は、[[神経変性]]による起こる[[認知症]]で、高齢化により患者数は増加している。アルツハイマー病の病態に過剰なグルタミン酸受容体の活性化が関与することは、グルタミン酸受容体阻害剤であるメマンチンが治療薬として用いられていることからも明らかである。グルタミン酸トランスポーターの障害がアルツハイマー病の発症に関与することを示す証拠として以下のものがある。 | ||
#アルツハイマー病患者の脳ではslc1a1、slc12、slc1a3の発現量が減少している<ref name=ref65><pubmed>21743130</pubmed></ref>。 | |||
#アルツハイマー病モデルのslc1a2発現量を低下させると空間学習の障害が促進される<ref name=ref66><pubmed>21677376</pubmed></ref> | |||
# | #アルツハイマー病における神経変性の原因物質と考えられている[[βアミロイド]]タンパク質によりslc1a2の機能が障害される<ref name=ref67><pubmed>23516295</pubmed></ref>。 | ||
# | #slc1a2の発現量を増加させる[[セフトリアキソン]]はアルツハイマー病モデルの異常を回復させる<ref name=ref68><pubmed>25964214</pubmed></ref> <ref name=ref69><pubmed>25711212</pubmed></ref>。 | ||
===筋萎縮性側索硬化症=== | ===筋萎縮性側索硬化症=== | ||
[[筋萎縮性側索硬化症]] | [[筋萎縮性側索硬化症]]は、筋肉の動きを制御する運動神経が選択的に変性する疾患である。筋萎縮性側索硬化症の病態に過剰なグルタミン酸受容体の活性化が関与することは、グルタミン酸シナプス伝達の阻害作用を持つリルゾールが治療薬として用いられていることからも明らかである。グルタミン酸トランスポーターの障害が筋萎縮性側索硬化症の発症に関与することを示す証拠として以下のものがある。 | ||
# | #筋萎縮性側索硬化症患者の脳脊髄液中のグルタミン酸濃度が増加している<ref name=ref70><pubmed>11790386</pubmed></ref>。 | ||
#筋萎縮性側索硬化症患者の脊髄においてグルタミン酸取り込み能とslc1a2の発現量が減少している | #筋萎縮性側索硬化症患者の脊髄においてグルタミン酸取り込み能とslc1a2の発現量が減少している<ref name=ref71><pubmed>1349424</pubmed></ref> <ref name=ref72><pubmed>7611729</pubmed></ref>。 | ||
# | #筋萎縮性側索硬化症モデル動物においてslc1a2とslc1a3の発編量が減少している<ref name=ref73><pubmed>11818550</pubmed></ref> <ref name=ref74><pubmed>23714777</pubmed></ref>。 | ||
#slc1a2を活性化する化合物は筋萎縮性側索硬化症モデルの症状を改善する | #slc1a2を活性化する化合物は筋萎縮性側索硬化症モデルの症状を改善する<ref name=ref61 /> <ref name=ref75><pubmed>15635412</pubmed></ref>。 | ||
===ハンチントン病=== | ===ハンチントン病=== | ||
[[ハンチントン病]] | [[ハンチントン病]]は、線条体の神経細胞が変性し、不随意運動・認知障害などの症状を示す常染色体優性遺伝疾患である。グルタミン酸トランスポーターの障害がハンチントン病の発症に関与することを示す証拠として以下のものがある。 | ||
#ハンチントン病患者の線条体においてslc1a2の発現が減少している | #ハンチントン病患者の線条体においてslc1a2の発現が減少している<ref name=ref76><pubmed>9100675</pubmed></ref> <ref name=ref77><pubmed>20494921</pubmed></ref>。 | ||
# | #ハンチントン病モデル動物においてslc1a2とslc1a3の発現量が減少している<ref name=ref77 /> <ref name=ref78><pubmed>19168136</pubmed></ref>。 | ||
#slc1a2を活性化する化合物はハンチントン病モデルの症状を改善する | #slc1a2を活性化する化合物はハンチントン病モデルの症状を改善する<ref name=ref79><pubmed>18353560</pubmed></ref>。 | ||
===片頭痛=== | ===片頭痛=== | ||
[[片頭痛]]を伴う[[エピソード性運動失調6型]]([[Episodic ataxia with hemiplegic migraine and seizures]])の患者及び弧発性片麻痺性片頭痛の患者に、slc1a3の機能を障害するミスセンス変異(C186S とP290R、T387P)が見つかっている<ref name=ref57 /> <ref name=ref58 /> <ref name=ref80>'''Freilinger T, Koch J, Dichgans M, Mamsa H, Jen JC.'''<br>A novel mutation in slc1a3 associated with pure hemiplegic migraine. <br>''J Headache Pain 11''(Suppl 1): S90, 2010.</ref>。純粋な片頭痛のみを示す患者にslc1a2の変異は見つかっていないが、slc1a2の機能・発現に関与する[[ATP1A2]]、[[slc4a4]]、[[MTDH]]の変異が報告されている<ref name=ref81><pubmed>12539047</pubmed></ref> <ref name=ref82><pubmed>20798035</pubmed></ref> <ref name=ref83><pubmed>20802479</pubmed></ref>。 | |||
===多発性硬化症=== | ===多発性硬化症=== | ||
[[多発性硬化症]]は、中枢神経系の脱髄疾患の一つである。グルタミン酸トランスポーターの障害が多発性硬化症の発症に関与することを示す証拠として以下のものがある。 | [[多発性硬化症]]は、中枢神経系の脱髄疾患の一つである。グルタミン酸トランスポーターの障害が多発性硬化症の発症に関与することを示す証拠として以下のものがある。 | ||
#多発性硬化症患者の脳内および脳脊髄液中のグルタミン酸濃度が増加している | #多発性硬化症患者の脳内および脳脊髄液中のグルタミン酸濃度が増加している<ref name=ref84><pubmed>15758036</pubmed></ref> <ref name=ref85><pubmed>23613944</pubmed></ref> <ref name=ref86><pubmed>9466133</pubmed></ref>。 | ||
# | #多発性硬化症患者の大脳皮質の障害部位ではslc1a2とslc1a3の発現が減少している<ref name=ref87><pubmed>17882017</pubmed></ref>。 | ||
#グルタミン酸受容体の阻害剤が多発性硬化症モデルの症状を改善する | #グルタミン酸受容体の阻害剤が多発性硬化症モデルの症状を改善する<ref name=ref88><pubmed>10613825</pubmed></ref> <ref name=ref89><pubmed>10613826</pubmed></ref>。 | ||
===本態性振戦=== | ===本態性振戦=== | ||
[[本態性振戦]]患者において、slc1a2の1塩基多型との関連や小脳でのslc1a2の発現減少が報告されている<ref name=ref90><pubmed>22764253</pubmed></ref> <ref name=ref91><pubmed>25391854</pubmed></ref>。 | |||
===脳梗塞=== | ===脳梗塞=== | ||
[[脳梗塞]]の障害程度は、slc1a2の発現量を規定する1塩基多型と相関することが報告されている<ref name=ref92><pubmed>16520390</pubmed></ref>。また、slc1a2欠損マウスでは[[虚血]]による障害が悪化し<ref name=ref93><pubmed>12904478</pubmed></ref>、slc1a2の活性化化合物の投与により虚血障害が軽度になる<ref name=ref94><pubmed>23523747</pubmed></ref> <ref name=ref95><pubmed>25270764</pubmed></ref>。 | |||
===慢性疼痛=== | ===慢性疼痛=== | ||
[[慢性疼痛]]モデルでのslc1a2の発現減少<ref name=ref96><pubmed>16242033</pubmed></ref>やslc1a2の活性化化合物が慢性疼痛モデルの疼痛を緩和する<ref name=ref97><pubmed>20022427</pubmed></ref>という報告があり、slc1a2が慢性疼痛の創薬の一つの標的になっている<ref name=ref98><pubmed>25270665</pubmed></ref>。 | |||
===緑内障=== | ===緑内障=== | ||
[[緑内障]]は、40歳以上では約5%が潜在的に罹患していると考えられており、日本人の中途失明原因の第1位である。さらに、高齢化により患者数は増加し、その治療は活力ある高齢化社会を作るためには必要不可欠である。我が国の緑内障の約70%は[[正常眼圧緑内障]]であり、その病態は不明である。グルタミン酸トランスポーター slc1a3欠損マウスは、眼圧が正常であるにも関わらず、[[網膜神経節細胞]]が加齢に伴い選択的に変性し、視神経乳頭陥凹が拡大するなど正常眼圧緑内障に似た症状を示す<ref name=ref99><pubmed>17607354</pubmed></ref>。また、slc1a3の発現を増加させる[[アルンジン酸]]はslic1a3欠損マウスの緑内障様症状を改善する<ref name=ref100><pubmed>25789968</pubmed></ref>。しかし、slc1a3の遺伝子変異が正常眼圧緑内障の直接的な原因になるかは不明である。 | |||
==参考文献== | ==参考文献== | ||
<references /> | <references /> | ||
2020年1月9日 (木) 11:27時点における最新版
田中 光一
東京医科歯科大学
DOI:10.14931/bsd.6750 原稿受付日:2016年1月26日 原稿完成日:2016年2月12日
担当編集委員:和田 圭司(国立研究開発法人国立精神・神経医療研究センター)
グルタミン酸は哺乳類の中枢神経系において記憶・学習などの高次機能を調節する主要な興奮性神経伝達物質として知られている。一方、過剰なグルタミン酸は、グルタミン酸受容体の過剰な活性化によりグルタミン酸興奮毒性と呼ばれる神経細胞障害作用を持つことが知られている。このため細胞外グルタミン酸濃度は厳密に制御される必要があり、グルタミン酸トランスポーターがその役割を担う。これまで哺乳類の中枢神経系において、5種類のグルタミン酸トランスポーターサブファミリー、slc1a1、slc1a2、slc1a3、slc1a6、slc1a7が単離されている。slc1a2、slc1a3は主にアストロサイトに、slc1a1とslc1a6は神経細胞に、slc1a7は網膜に発現している。シナプス間隙におけるグルタミン酸の除去は、主にアストロサイトに存在する2種類のグルタミン酸輸送体slc1a2、slc1a3により担われている。近年、これらのグルタミン酸トランスポーターの機能障害が様々な精神神経疾患の発症に関与することが明らかになりつつある。
| グルタミン酸トランスポーター | |||||||||
|---|---|---|---|---|---|---|---|---|---|
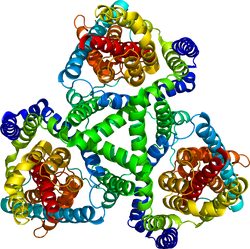 グルタミン酸トランスポーターホモログ(Pyrococcus horikoshii) | |||||||||
| Identifiers | |||||||||
| Symbol | ? | ||||||||
| Pfam | PF00375 | ||||||||
| InterPro | IPR001991 | ||||||||
| PROSITE | PDOC00591 | ||||||||
| SCOP | 1XFH | ||||||||
| SUPERFAMILY | 1XFH | ||||||||
| TCDB | 2.A.23.1.5 | ||||||||
| OPM protein | 2nwl | ||||||||
| |||||||||
グルタミン酸トランスポーターとは

Glu:グルタミン酸、Gln:グルタミン、GS:グルタミン合成酵素、GT:グルタミナーゼ

グルタミン酸は、哺乳類中枢神経系において約70%の神経細胞が用いる主要な興奮性神経伝達物質であり、記憶・学習などの脳高次機能に重要な役割を果たしている[1]。しかし、その機能的な重要性の反面、興奮毒性という概念で表されるように[2]、過剰なグルタミン酸は神経細胞障害作用を持ち、様々な精神神経疾患の発症に関与することが明らかになりつつある[3] [4]。従って、シナプス間隙におけるグルタミン酸濃度は厳密に制御されなければならない。
シナプスにおけるグルタミン酸の動態は図1のように考えられている。シナプス前終末から放出されたグルタミン酸は、シナプス後細胞のグルタミン酸受容体に結合しその効果を発揮するが、伝達終了後シナプス間隙のグルタミン酸はアストロサイトおよびシナプス後神経細胞膜に存在するグルタミン酸トランスポーターにより細胞内に取り込まれる。アストロサイトに取り込まれたグルタミン酸は、グルタミン合成酵素によりグルタミンに変換され、グリア細胞外に放出され、グルタミン-グルタミン酸サイクルを経て、再びシナプス小胞に蓄えられる。
中枢神経系には5種類のグルタミン酸トランスポーターサブファミリーが存在することが知られおり[5]、グルタミン酸トランスポーター欠損マウスの解析を通じ、グルタミン酸トランスポーターの各サブファミリーの機能的役割が明らかになりつつある。
構造
5種類のグルタミン酸トランスポーターは互いに50~55%の相同性を有するアミノ酸数520~580のタンパク質である。グルタミン酸トランスポーターの構造はN末端とC末端は細胞内に位置し、8個の膜貫通部位、2個の膜を完全に貫通していないループ構造(re-entrant loop)を持つ(図2)[6]。グルタミン酸トランスポーターはグルタミン酸を取り込む際に、Na+およびH+の共輸送、K+の対向輸送と共役する。グルタミン酸、Na+、H+、K+の結合・輸送に関与しているアミノ酸残基はいずれも6番目の膜貫通領域以降のC末端側である。また、グルタミン酸トランスポーターは3量体のホモオリゴマーとして細胞膜に発現している。
サブファミリー
グルタミン酸トランスポーターには、slc1a1、slc1a2、slc1a3、slc1a6、slc1a7の5種類のサブファミリーが知られている(表1)。
| 別の名称 | アミノ酸残基数(ヒト) | 組織分布 | 中枢神経系での細胞局在 | 遺伝子座(ヒト) | |
| slc1a1 | EAAT3 EAAC1 |
524 | 脳・心臓・腎臓・肝臓・小腸 | 神経細胞 | 9p24 |
| slc1a2 | EAAT2 GLT-1 |
574 | 脳・肝臓・膵臓 | アストロサイト | 11p13-p12 |
| slc1a3 | EAAT1 GLAST |
542 | 脳・心臓・肺・骨格筋・胎盤 | アストロサイト | 5p13 |
| slc1a6 | EAAT4 | 564 | 脳・胎盤 | プルキンエ細胞 | 19p13.12 |
| slc1a7 | EAAT5 | 560 | 網膜・肝臓・心臓・肺・骨格筋・小腸 | 視細胞・双極細胞 | 1p32.3 |
発現

5種類のグルタミン酸トランスポーターサブファミリーは、中枢神経系だけでなく末梢組織にも発現していることが知られているが、本稿では中枢神経系における発現に関して記述する(表1)。
slc1a2(GLT-1/EAAT2)は大脳皮質・海馬のアストロサイトに、slc1a3(GLAST/EAAT1)は小脳のアストロサイトに優位に発現している[7]。slc1a1(EAAC1/EAAT3)は神経細胞に存在し、中枢神経系に広く分布している[7]。slc1a6(EAAT4)は小脳のプルキンエ細胞に[8]、またslc1a7(EAAT5)は網膜の視細胞・双極細胞に特異的に発現している[9](図3)。神経細胞に発現しているslc1a1とslc1a4は、神経細胞の終末ではなく細胞体・樹状突起に主に局在している[10] [11]。アストロサイトに発現しているslc1a2とslc1a3は、シナプス周囲を覆っている突起に密度高く局在している[12]。
最近、CDC42EP4/septinがslc1a3をシナプス周囲を覆うバーグマングリアの突起に局在させることが明らかになった[13]。成人脳ではアストロサイトに局在するslc1a2は、胎児期から生後3日の発生初期には、一過性に神経細胞に発現する[14]。
また、成人脳ではアストロサイトに局在するslc1a3は、発生初期には神経幹細胞に発現しており、神経幹細胞のマーカーとして用いられている[15] [16]。
機能

分子機能
グルタミン酸トランスポーターは、細胞膜を介したNa+の電気化学ポテンシャルを利用して、グルタミン酸を輸送する。1分子のグルタミン酸の取り込みは、3個のNa+および1個のH+の共輸送、1個のK+の対向輸送と共役する(図2)。従って、グルタミン酸トランスポーターは起電性であり、グルタミン酸の細胞内への取り込みにより内向き電流が生じる。また、これとは別に、熱力学的にグルタミン酸取り込みと連動していないCl-の流入があることが知られているが、Cl-の透過性の順番はslc1a6/7 > slc1a3 > slc1a1 > slc1a2である[17]。
slc1a1は、グルタミン酸の他に電荷をもたないL-システインを取り込み、グルタチオン合成に利用している[18]。
生理機能
海馬のシナプス伝達における役割
海馬において主要なグルタミン酸トランスポーターはslc1a2である。slc1a2欠損マウスの海馬のシェーファー側枝・CA1錐体細胞間シナプスを電気生理学的に調べたところ、海馬のCA1錐体細胞で記録されるシェーファー側枝による興奮性シナプス後電流(Excitatory Postsynaptic Current:EPSC)のAMPA(α-amino-3-hydroxy-5-methylisoxazole-4- propionic acid)型グルタミン酸受容体成分・NMDA型グルタミン酸受容体成分とも、その振幅・時間経過は野生型と違いはなかった[19]。このことは、GLT1は海馬において、EPSCの振幅・時間経過の重要な決定因子ではないことを示している。海馬におけるEPSCの振幅・時間経過は、グルタミン酸受容体自体のキネテクスにより規定されていると考えられる。海馬のシナプスは、グリア細胞の突起によるシナプス部位の被覆が不完全で、主に拡散がシナプス間隙におけるグルタミン酸のクリアランスを規定していると考えられる。
海馬CA1網状分子層のoriens-lacunosum moleculare(O-LM)介在神経に多く存在する代謝型グルタミン酸受容体mGluR1は、グリア型グルタミン酸トランスポーターslc1a2およびslc1a3により、活性が制御されている。slc1a2とslc1a3を抑制すると、mGluR1依存性EPSCの振幅が増加し、介在神経の発火が増強され、結果としてCA1錐体細胞の抑制が増強されたる[20]。
また、slc1a2欠損マウスでは、海馬CA1領域のNMDA型グルタミン酸受容体成分が増強され、長期増強の発現が障害されている[21]。逆に、slc1a2の発現を増加すると、苔状線維—CA3錐体細胞間シナプスの長期抑圧の発現が障害される[22]。
小脳のシナプス伝達における役割
小脳ではプルキンエ細胞にグルタミン酸トランスポーターslc1a1、slc1a6が、プルキンエ細胞を取り囲むバーグマングリア細胞にはslc1a2, slc1a3が発現している。欠損マウスの解析から、平行線維・プルキンエ細胞間シナプス、登上線維・プルキンエ細胞間シナプスにおいて、グルタミン酸が放出された直後の高濃度のグルタミン酸の除去はslc1a3により、放出されてしばらく時間が経過した後の低濃度のグルタミン酸の除去はslc1a6により行われていることがわかった[23] [24]。さらにslc1a1とslc1a6の選択的機能阻害により隣接するシナプスへのグルタミン酸spilloverが起こり、mGluR1依存性EPSCが増強、長期抑圧が促進される[25] [26]。また、slc1a6は、平行線維・バーグマングリア間のグルタミン酸伝達を制御している[27]。
大脳皮質のシナプス伝達の維持における役割
神経系は他の臓器に比べエネルギー要求性が高く、そのほとんどはシナプス伝達に使われる。従って、シナプス伝達を維持するためには、活動の亢進した部位に選択的にエネルギーを補充する必要がある。グリア型グルタミン酸トランスポーターslc1a2・slc1a3は、シナプス伝達のセンサーとして働き、神経活動の亢進→シナプス間隙のグルタミン酸濃度上昇→グリア型グルタミン酸トランスポーターによるグルタミン酸の再吸収(同時にNa+がグリア内へ流入)→グリアのNa+-K+ ATPaseの活性化(グリア内でのエネルギー消費増大)→グリアによる毛細血管からのブドウ糖の取り込み増加→グリアの解糖系によるブドウ糖から乳酸の生成(グリア内の消費したエネルギーの補充)→生成した乳酸を神経細胞が取り込みエネルギーを補充、という一連のエネルギー補給反応をトリガーする(図4)[28] [29]。
リボンシナプスにおける役割
リボンシナプスでは、シナプス前細胞は活動電位を出さず、代わりに膜電位を連続的に変化させることで伝達物質の放出量を変化させ、情報を伝達する。リボンシナプスは網膜や内耳などの一次知覚のシナプスに存在する。
聴覚の一次知覚シナプスである内有毛細胞—蝸牛神経間シナプスは典型的なリボンシナプスであり、グルタミン酸が神経伝達物質である。内有毛細胞周囲の支持細胞(inner phalangeal cell, IPC)にはslc1a3が、 蝸牛神経節細胞にはslc1a1とslc1a2が存在する。欠損マウスや薬理学的解析の結果から、内有毛細胞—蝸牛神経間シナプスのグルタミン酸の除去はIPCに存在するslc1a3により行われていることが明らかになった[30] [31]
また、典型的なリボンシナプスの一つである網膜の視細胞—双極細胞間シナプスでは、視細胞に存在するグルタミン酸トランスポーターslc1a7がシナプス間隙からのグルタミン酸除去に主要な役割を果たす[32]。
脳形成における役割
胎児期の脳には、slc1a1、slc1a2、slc1a3の3種類のグルタミン酸トランスポーターが存在する。これら3種類のサブファミリーの単独欠損マウスおよびslc1a1とslc1a3、slc1a1とslc1a2ダブル欠損マウスは正常な脳形成を示すが、slc1a2とslc1a3ダブル欠損マウスは胎生17日頃に死亡し、海馬・大脳皮質に層形成異常や大脳皮質と他の脳部位を結合する線維連絡に異常が観察される[33]。これらの異常はNMDA型グルタミン酸受容体サブユニット1の欠損により改善する[34]。細胞外に過剰に存在するグルタミン酸は、NMDA型グルタミン酸受容体を過剰に活性化し、神経細胞の移動、神経幹細胞の分裂、神経突起の伸長を障害する。従って、脳が正常に発達するには、slc1a2とslc1a3による細胞外グルタミン酸濃度の厳密な制御が必要である。
疾患との関わり

脳の興奮性亢進は、多くの精神神経疾患に共通する病態と考えられている(図5)。脳の興奮性を亢進させる原因として、グルタミン酸シナプス伝達の亢進とGABAシナプス伝達の低下がある。グルタミン酸トランスポーターの機能障害は、グルタミン酸シナプス伝達を亢進させる主要な原因の一つである。
統合失調症
統合失調症は、幻聴・妄想などの陽性症状と、感情鈍麻、意欲の減退などの陰性症状、作業記憶などの認知障害を示し、有病率約1%の精神疾患である。統合失調症患者の遺伝子解析から、slc1a3遺伝子座の欠失やslc1a2の機能障害を伴うミスセンス変異を持つ症例が報告された[35] [36]。また、死後脳の解析からも、slc1a2およびslc1a3の発現が減少することが報告されている[37]。slc1a3欠損マウスは、統合失調症の陽性症状・陰性症状・認知障害に相当する行動異常を示し、slc1a3の異常による脳の興奮性亢進が統合失調症の発症に重要な役割を果たすと考えられる[38] [39]。さらに、統合失調症の前駆期から初発期への移行に細胞外グルタミン酸濃度の上昇が関与することが報告されている[40] [41]。
うつ病
うつ病は、抑うつ気分と興味・喜びの喪失などを特徴とする精神疾患である。近年、うつ病においても、以下のようなグルタミン酸神経伝達の亢進を示唆する知見が報告され、一つの仮説を形成しつつある[42] [43]。
- NMDA型グルタミン酸受容体阻害剤であるケタミンが、うつ病患者に即効性の抗うつ作用を示す[42]。
- うつ病患者の血中・脳脊髄液中・脳内のグルタミン酸濃度は上昇している[44] [45]。
- うつ病患者の死後脳ではslc1a2とslc1a3の発現が減少している[46]。
- slc1a2の発現を増加させるβ-ラクタム系抗生物質が、マウスのうつ様行動を改善する[47]。
- slc1a2とslc1a3を活性化するリルゾールは、うつ病に効果がある[48]。
手綱核特異的slc1a2欠損マウスは、うつ病の症状に似た行動異常や睡眠障害を起こす[49]。これらの知見は、グルタミン酸トランスポーターの機能不全によるグルタミン酸神経伝達の過剰な活性化が、うつ病の発症に重要な役割を果たしていることを示唆している。
双極性障害
双極性障害は、うつ状態と躁状態を繰り返す慢性の疾患である。1H-MRS(magnetic resonance spectroscopy)によるグルタミン酸濃度測定のメタ解析で、双極性障害の患者において脳のグルタミン酸濃度が増加していることが報告されている[50]。また、患者死後脳におけるslc1a2の発現減少や、双極性障害患者にのみ見られるslc1a2の稀なミスセンス変異(G6S、A20S、R31Q)が報告されている[51]。さらに、NMDA型グルタミン酸受容体の阻害剤であるメマンチンが躁病エピソードの治療に効果があるという報告もなされている[52]。
強迫性障害
強迫性障害は、強迫観念・強迫行為を特徴とする疾患である。多くは思春期過ぎから発症し、人口の2~3%が罹患歴を持つ。近年、強迫性障害においても、以下のようなグルタミン酸神経伝達の亢進を示唆する知見が報告されている。
- 強迫性障害患者の脳内ではグルタミン酸濃度が増加し、神経伝達が亢進している[53]。
- slc1a3の一塩基多型と強迫性障害の関連が報告されている[54]。
- グルタミン酸神経伝達を抑制する薬剤に強迫性障害の治療効果がある[53]。
自閉スペクトラム症
自閉スペクトラム症は、社会性行動の喪失・言語発達の遅延・限局した興味や繰り返し行動を特徴とする脳高次機能の発達障害である。
自閉症様の行動を示す脆弱X症候群や結節性硬化症の患者ではグルタミン酸神経伝達の亢進が報告されている。自閉症のゲノムワイドな連鎖解析により、11番染色体の11p12-13が自閉症に関連があることが明らかになった[55]。この領域はslc1a2の遺伝子座の近傍である。また、slc1a2を性成熟期以降に欠損させたマウスでは、大脳皮質—線条体間のグルタミン酸伝達が過剰に活性化され、過度な毛繕い行動と突発的に全身を激しく震わせるwet-dog shakeと呼ばれる繰り返し行動の回数が大幅に増加することが報告されている[56]。
てんかん
てんかんは、ニューロンの過剰な活動に伴い痙攣や意識障害などが発作的に反復して起こる慢性的な脳疾患である。てんかん発作の発現機序として、脳内の抑制性と興奮性神経伝達の不均衡状態が重要だと考えられている。内側側頭葉てんかん患者の海馬において、細胞外グルタミン酸濃度が上昇することが報告されている。しかし、側頭葉てんかん患者の外科切除海馬標本を用いた解析では、グルタミン酸トランスポーターslc1a2とslc1a3の発現減少に関して統一した結果が得られていない。グルタミン酸トランスポーターの遺伝子解析では、てんかん発作を伴うエピソード性運動失調6型(Episodic ataxia with hemiplegic migraine and seizures)の患者にslc1a3の機能を障害するミスセンス変異(C186S とP290R)が見つかっている[57] [58]。
また、slc1a2欠損マウスは、致死性の自発てんかん発作により、生後3週齢から突然死を起こす。てんかん発作パターンは、NMDAを皮下注した時に見られる発作と類似しており、突然ケージの中を走り回り反弓緊張様姿勢をとり死亡する[19]。さらに、slc1a3欠損マウスは、自発性てんかん発作は観察されないが、pentylentetorazole(PTZ)誘発性てんかんに対する感受性が亢進している[59]。また、slic1a2の発現を増加させる薬物は、様々なてんかんモデルにおいて、抗てんかん作用を示すことが報告されている[60] [61]。
薬物依存
モルヒネ、コカイン、覚せい剤、アルコールなどの依存性薬物は、その反復摂取により依存性が形成される。近年の薬物乱用の低年齢化や一般市民への拡大は、社会的にも大きな問題となっている。薬物依存の形成・維持・再燃過程に、グルタミン酸神経伝達の亢進が重要な役割を果たすことは、以下の知見から示唆されている[62] [63] [64]。
- 薬物依存モデルの脳において細胞外グルタミン酸濃度が増加する。
- 薬物依存モデルの脳においてslc1a2の発現量が減少する。
- slc1a2の阻害剤により、薬物依存の形成・維持・再燃過程が増悪する。
- slc1a2の活性化薬や過剰発現により、薬物依存の形成・維持・再燃過程が減弱する。
アルツハイマー病
アルツハイマー病は、神経変性による起こる認知症で、高齢化により患者数は増加している。アルツハイマー病の病態に過剰なグルタミン酸受容体の活性化が関与することは、グルタミン酸受容体阻害剤であるメマンチンが治療薬として用いられていることからも明らかである。グルタミン酸トランスポーターの障害がアルツハイマー病の発症に関与することを示す証拠として以下のものがある。
- アルツハイマー病患者の脳ではslc1a1、slc12、slc1a3の発現量が減少している[65]。
- アルツハイマー病モデルのslc1a2発現量を低下させると空間学習の障害が促進される[66]
- アルツハイマー病における神経変性の原因物質と考えられているβアミロイドタンパク質によりslc1a2の機能が障害される[67]。
- slc1a2の発現量を増加させるセフトリアキソンはアルツハイマー病モデルの異常を回復させる[68] [69]。
筋萎縮性側索硬化症
筋萎縮性側索硬化症は、筋肉の動きを制御する運動神経が選択的に変性する疾患である。筋萎縮性側索硬化症の病態に過剰なグルタミン酸受容体の活性化が関与することは、グルタミン酸シナプス伝達の阻害作用を持つリルゾールが治療薬として用いられていることからも明らかである。グルタミン酸トランスポーターの障害が筋萎縮性側索硬化症の発症に関与することを示す証拠として以下のものがある。
- 筋萎縮性側索硬化症患者の脳脊髄液中のグルタミン酸濃度が増加している[70]。
- 筋萎縮性側索硬化症患者の脊髄においてグルタミン酸取り込み能とslc1a2の発現量が減少している[71] [72]。
- 筋萎縮性側索硬化症モデル動物においてslc1a2とslc1a3の発編量が減少している[73] [74]。
- slc1a2を活性化する化合物は筋萎縮性側索硬化症モデルの症状を改善する[61] [75]。
ハンチントン病
ハンチントン病は、線条体の神経細胞が変性し、不随意運動・認知障害などの症状を示す常染色体優性遺伝疾患である。グルタミン酸トランスポーターの障害がハンチントン病の発症に関与することを示す証拠として以下のものがある。
- ハンチントン病患者の線条体においてslc1a2の発現が減少している[76] [77]。
- ハンチントン病モデル動物においてslc1a2とslc1a3の発現量が減少している[77] [78]。
- slc1a2を活性化する化合物はハンチントン病モデルの症状を改善する[79]。
片頭痛
片頭痛を伴うエピソード性運動失調6型(Episodic ataxia with hemiplegic migraine and seizures)の患者及び弧発性片麻痺性片頭痛の患者に、slc1a3の機能を障害するミスセンス変異(C186S とP290R、T387P)が見つかっている[57] [58] [80]。純粋な片頭痛のみを示す患者にslc1a2の変異は見つかっていないが、slc1a2の機能・発現に関与するATP1A2、slc4a4、MTDHの変異が報告されている[81] [82] [83]。
多発性硬化症
多発性硬化症は、中枢神経系の脱髄疾患の一つである。グルタミン酸トランスポーターの障害が多発性硬化症の発症に関与することを示す証拠として以下のものがある。
- 多発性硬化症患者の脳内および脳脊髄液中のグルタミン酸濃度が増加している[84] [85] [86]。
- 多発性硬化症患者の大脳皮質の障害部位ではslc1a2とslc1a3の発現が減少している[87]。
- グルタミン酸受容体の阻害剤が多発性硬化症モデルの症状を改善する[88] [89]。
本態性振戦
本態性振戦患者において、slc1a2の1塩基多型との関連や小脳でのslc1a2の発現減少が報告されている[90] [91]。
脳梗塞
脳梗塞の障害程度は、slc1a2の発現量を規定する1塩基多型と相関することが報告されている[92]。また、slc1a2欠損マウスでは虚血による障害が悪化し[93]、slc1a2の活性化化合物の投与により虚血障害が軽度になる[94] [95]。
慢性疼痛
慢性疼痛モデルでのslc1a2の発現減少[96]やslc1a2の活性化化合物が慢性疼痛モデルの疼痛を緩和する[97]という報告があり、slc1a2が慢性疼痛の創薬の一つの標的になっている[98]。
緑内障
緑内障は、40歳以上では約5%が潜在的に罹患していると考えられており、日本人の中途失明原因の第1位である。さらに、高齢化により患者数は増加し、その治療は活力ある高齢化社会を作るためには必要不可欠である。我が国の緑内障の約70%は正常眼圧緑内障であり、その病態は不明である。グルタミン酸トランスポーター slc1a3欠損マウスは、眼圧が正常であるにも関わらず、網膜神経節細胞が加齢に伴い選択的に変性し、視神経乳頭陥凹が拡大するなど正常眼圧緑内障に似た症状を示す[99]。また、slc1a3の発現を増加させるアルンジン酸はslic1a3欠損マウスの緑内障様症状を改善する[100]。しかし、slc1a3の遺伝子変異が正常眼圧緑内障の直接的な原因になるかは不明である。
参考文献
- ↑
Nakanishi, S., Nakajima, Y., Masu, M., Ueda, Y., Nakahara, K., Watanabe, D., ..., & Okada, M. (1998).
Glutamate receptors: brain function and signal transduction. Brain research. Brain research reviews, 26(2-3), 230-5. [PubMed:9651535] - ↑
Choi, D.W. (1988).
Glutamate neurotoxicity and diseases of the nervous system. Neuron, 1(8), 623-34. [PubMed:2908446] [WorldCat] [DOI] - ↑
Miladinovic, T., Nashed, M.G., & Singh, G. (2015).
Overview of Glutamatergic Dysregulation in Central Pathologies. Biomolecules, 5(4), 3112-41. [PubMed:26569330] [PMC] [WorldCat] [DOI] - ↑
McCullumsmith, R.E., & Sanacora, G. (2015).
Regulation of extrasynaptic glutamate levels as a pathophysiological mechanism in disorders of motivation and addiction. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology, 40(1), 254-5. [PubMed:25482181] [PMC] [WorldCat] [DOI] - ↑
Danbolt, N.C. (2001).
Glutamate uptake. Progress in neurobiology, 65(1), 1-105. [PubMed:11369436] [WorldCat] - ↑
Jiang, J., & Amara, S.G. (2011).
New views of glutamate transporter structure and function: advances and challenges. Neuropharmacology, 60(1), 172-81. [PubMed:20708631] [PMC] [WorldCat] [DOI] - ↑ 7.0 7.1
Shibata, T., Watanabe, M., Tanaka, K., Wada, K., & Inoue, Y. (1996).
Dynamic changes in expression of glutamate transporter mRNAs in developing brain. Neuroreport, 7(3), 705-9. [PubMed:8733726] [WorldCat] [DOI] - ↑
Yamada, K., Watanabe, M., Shibata, T., Tanaka, K., Wada, K., & Inoue, Y. (1996).
EAAT4 is a post-synaptic glutamate transporter at Purkinje cell synapses. Neuroreport, 7(12), 2013-7. [PubMed:8905715] [WorldCat] [DOI] - ↑
Pow, D.V., & Barnett, N.L. (2000).
Developmental expression of excitatory amino acid transporter 5: a photoreceptor and bipolar cell glutamate transporter in rat retina. Neuroscience letters, 280(1), 21-4. [PubMed:10696802] [WorldCat] [DOI] - ↑
Rothstein, J.D., Martin, L., Levey, A.I., Dykes-Hoberg, M., Jin, L., Wu, D., ..., & Kuncl, R.W. (1994).
Localization of neuronal and glial glutamate transporters. Neuron, 13(3), 713-25. [PubMed:7917301] [WorldCat] [DOI] - ↑
Tanaka, J., Ichikawa, R., Watanabe, M., Tanaka, K., & Inoue, Y. (1997).
Extra-junctional localization of glutamate transporter EAAT4 at excitatory Purkinje cell synapses. Neuroreport, 8(11), 2461-4. [PubMed:9261809] [WorldCat] [DOI] - ↑
Chaudhry, F.A., Lehre, K.P., van Lookeren Campagne, M., Ottersen, O.P., Danbolt, N.C., & Storm-Mathisen, J. (1995).
Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron, 15(3), 711-20. [PubMed:7546749] [WorldCat] [DOI] - ↑
Ageta-Ishihara, N., Yamazaki, M., Konno, K., Nakayama, H., Abe, M., Hashimoto, K., ..., & Kinoshita, M. (2015).
A CDC42EP4/septin-based perisynaptic glial scaffold facilitates glutamate clearance. Nature communications, 6, 10090. [PubMed:26657011] [PMC] [WorldCat] [DOI] - ↑
Yamada, K., Watanabe, M., Shibata, T., Nagashima, M., Tanaka, K., & Inoue, Y. (1998).
Glutamate transporter GLT-1 is transiently localized on growing axons of the mouse spinal cord before establishing astrocytic expression. The Journal of neuroscience : the official journal of the Society for Neuroscience, 18(15), 5706-13. [PubMed:9671661] [PMC] [WorldCat] - ↑
Shibata, T., Yamada, K., Watanabe, M., Ikenaka, K., Wada, K., Tanaka, K., & Inoue, Y. (1997).
Glutamate transporter GLAST is expressed in the radial glia-astrocyte lineage of developing mouse spinal cord. The Journal of neuroscience : the official journal of the Society for Neuroscience, 17(23), 9212-9. [PubMed:9364068] [WorldCat] - ↑
Yang, S.M., Alvarez, D.D., & Schinder, A.F. (2015).
Reliable Genetic Labeling of Adult-Born Dentate Granule Cells Using Ascl1 CreERT2 and Glast CreERT2 Murine Lines. The Journal of neuroscience : the official journal of the Society for Neuroscience, 35(46), 15379-90. [PubMed:26586824] [PMC] [WorldCat] [DOI] - ↑
Cater, R.J., Ryan, R.M., & Vandenberg, R.J. (2016).
The Split Personality of Glutamate Transporters: A Chloride Channel and a Transporter. Neurochemical research, 41(3), 593-9. [PubMed:26303507] [WorldCat] [DOI] - ↑
Watts, S.D., Torres-Salazar, D., Divito, C.B., & Amara, S.G. (2014).
Cysteine transport through excitatory amino acid transporter 3 (EAAT3). PloS one, 9(10), e109245. [PubMed:25275463] [PMC] [WorldCat] [DOI] - ↑ 19.0 19.1
Tanaka, K., Watase, K., Manabe, T., Yamada, K., Watanabe, M., Takahashi, K., ..., & Wada, K. (1997).
Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science (New York, N.Y.), 276(5319), 1699-702. [PubMed:9180080] [WorldCat] [DOI] - ↑
Huang, Y.H., Sinha, S.R., Tanaka, K., Rothstein, J.D., & Bergles, D.E. (2004).
Astrocyte glutamate transporters regulate metabotropic glutamate receptor-mediated excitation of hippocampal interneurons. The Journal of neuroscience : the official journal of the Society for Neuroscience, 24(19), 4551-9. [PubMed:15140926] [PMC] [WorldCat] [DOI] - ↑
Katagiri, H., Tanaka, K., & Manabe, T. (2001).
Requirement of appropriate glutamate concentrations in the synaptic cleft for hippocampal LTP induction. The European journal of neuroscience, 14(3), 547-53. [PubMed:11553304] [WorldCat] [DOI] - ↑
Omrani, A., Melone, M., Bellesi, M., Safiulina, V., Aida, T., Tanaka, K., ..., & Conti, F. (2009).
Up-regulation of GLT-1 severely impairs LTD at mossy fibre--CA3 synapses. The Journal of physiology, 587(Pt 19), 4575-88. [PubMed:19651762] [PMC] [WorldCat] [DOI] - ↑
Takayasu, Y., Iino, M., Kakegawa, W., Maeno, H., Watase, K., Wada, K., ..., & Ozawa, S. (2005).
Differential roles of glial and neuronal glutamate transporters in Purkinje cell synapses. The Journal of neuroscience : the official journal of the Society for Neuroscience, 25(38), 8788-93. [PubMed:16177048] [PMC] [WorldCat] [DOI] - ↑
Takatsuru, Y., Takayasu, Y., Iino, M., Nikkuni, O., Ueda, Y., Tanaka, K., & Ozawa, S. (2006).
Roles of glial glutamate transporters in shaping EPSCs at the climbing fiber-Purkinje cell synapses. Neuroscience research, 54(2), 140-8. [PubMed:16377014] [WorldCat] [DOI] - ↑
Nikkuni, O., Takayasu, Y., Iino, M., Tanaka, K., & Ozawa, S. (2007).
Facilitated activation of metabotropic glutamate receptors in cerebellar Purkinje cells in glutamate transporter EAAT4-deficient mice. Neuroscience research, 59(3), 296-303. [PubMed:17727989] [WorldCat] [DOI] - ↑
Takayasu, Y., Iino, M., Takatsuru, Y., Tanaka, K., & Ozawa, S. (2009).
Functions of glutamate transporters in cerebellar Purkinje cell synapses. Acta physiologica (Oxford, England), 197(1), 1-12. [PubMed:19583702] [WorldCat] [DOI] - ↑
Tsai, M.C., Tanaka, K., Overstreet-Wadiche, L., & Wadiche, J.I. (2012).
Neuronal glutamate transporters regulate glial excitatory transmission. The Journal of neuroscience : the official journal of the Society for Neuroscience, 32(5), 1528-35. [PubMed:22302796] [PMC] [WorldCat] [DOI] - ↑
Voutsinos-Porche, B., Bonvento, G., Tanaka, K., Steiner, P., Welker, E., Chatton, J.Y., ..., & Pellerin, L. (2003).
Glial glutamate transporters mediate a functional metabolic crosstalk between neurons and astrocytes in the mouse developing cortex. Neuron, 37(2), 275-86. [PubMed:12546822] [WorldCat] [DOI] - ↑
Herard, A.S., Dubois, A., Escartin, C., Tanaka, K., Delzescaux, T., Hantraye, P., & Bonvento, G. (2005).
Decreased metabolic response to visual stimulation in the superior colliculus of mice lacking the glial glutamate transporter GLT-1. The European journal of neuroscience, 22(7), 1807-11. [PubMed:16197522] [WorldCat] [DOI] - ↑
Hakuba, N., Koga, K., Gyo, K., Usami, S.I., & Tanaka, K. (2000).
Exacerbation of noise-induced hearing loss in mice lacking the glutamate transporter GLAST. The Journal of neuroscience : the official journal of the Society for Neuroscience, 20(23), 8750-3. [PubMed:11102482] [PMC] [WorldCat] - ↑
Glowatzki, E., Cheng, N., Hiel, H., Yi, E., Tanaka, K., Ellis-Davies, G.C., ..., & Bergles, D.E. (2006).
The glutamate-aspartate transporter GLAST mediates glutamate uptake at inner hair cell afferent synapses in the mammalian cochlea. The Journal of neuroscience : the official journal of the Society for Neuroscience, 26(29), 7659-64. [PubMed:16855093] [PMC] [WorldCat] [DOI] - ↑
Hasegawa, J., Obara, T., Tanaka, K., & Tachibana, M. (2006).
High-density presynaptic transporters are required for glutamate removal from the first visual synapse. Neuron, 50(1), 63-74. [PubMed:16600856] [WorldCat] [DOI] - ↑
Matsugami, T.R., Tanemura, K., Mieda, M., Nakatomi, R., Yamada, K., Kondo, T., ..., & Tanaka, K. (2006).
From the Cover: Indispensability of the glutamate transporters GLAST and GLT1 to brain development. Proceedings of the National Academy of Sciences of the United States of America, 103(32), 12161-6. [PubMed:16880397] [PMC] [WorldCat] [DOI] - ↑
Aida, T., Ito, Y., Takahashi, Y.K., & Tanaka, K. (2012).
Overstimulation of NMDA receptors impairs early brain development in vivo. PloS one, 7(5), e36853. [PubMed:22606296] [PMC] [WorldCat] [DOI] - ↑
Walsh, T., McClellan, J.M., McCarthy, S.E., Addington, A.M., Pierce, S.B., Cooper, G.M., ..., & Sebat, J. (2008).
Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science (New York, N.Y.), 320(5875), 539-43. [PubMed:18369103] [WorldCat] [DOI] - ↑
Need, A.C., McEvoy, J.P., Gennarelli, M., Heinzen, E.L., Ge, D., Maia, J.M., ..., & Goldstein, D.B. (2012).
Exome sequencing followed by large-scale genotyping suggests a limited role for moderately rare risk factors of strong effect in schizophrenia. American journal of human genetics, 91(2), 303-12. [PubMed:22863191] [PMC] [WorldCat] [DOI] - ↑
Shan, D., Lucas, E.K., Drummond, J.B., Haroutunian, V., Meador-Woodruff, J.H., & McCullumsmith, R.E. (2013).
Abnormal expression of glutamate transporters in temporal lobe areas in elderly patients with schizophrenia. Schizophrenia research, 144(1-3), 1-8. [PubMed:23356950] [PMC] [WorldCat] [DOI] - ↑
Karlsson, R.M., Tanaka, K., Heilig, M., & Holmes, A. (2008).
Loss of glial glutamate and aspartate transporter (excitatory amino acid transporter 1) causes locomotor hyperactivity and exaggerated responses to psychotomimetics: rescue by haloperidol and metabotropic glutamate 2/3 agonist. Biological psychiatry, 64(9), 810-4. [PubMed:18550032] [PMC] [WorldCat] [DOI] - ↑
Karlsson, R.M., Tanaka, K., Saksida, L.M., Bussey, T.J., Heilig, M., & Holmes, A. (2009).
Assessment of glutamate transporter GLAST (EAAT1)-deficient mice for phenotypes relevant to the negative and executive/cognitive symptoms of schizophrenia. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology, 34(6), 1578-89. [PubMed:19078949] [PMC] [WorldCat] [DOI] - ↑
Kraguljac, N.V., White, D.M., Reid, M.A., & Lahti, A.C. (2013).
Increased hippocampal glutamate and volumetric deficits in unmedicated patients with schizophrenia. JAMA psychiatry, 70(12), 1294-302. [PubMed:24108440] [WorldCat] [DOI] - ↑
Schobel, S.A., Chaudhury, N.H., Khan, U.A., Paniagua, B., Styner, M.A., Asllani, I., ..., & Small, S.A. (2013).
Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron, 78(1), 81-93. [PubMed:23583108] [PMC] [WorldCat] [DOI] - ↑ 42.0 42.1
Sanacora, G., Zarate, C.A., Krystal, J.H., & Manji, H.K. (2008).
Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nature reviews. Drug discovery, 7(5), 426-37. [PubMed:18425072] [PMC] [WorldCat] [DOI] - ↑
Sanacora, G., Treccani, G., & Popoli, M. (2012).
Towards a glutamate hypothesis of depression: an emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology, 62(1), 63-77. [PubMed:21827775] [PMC] [WorldCat] [DOI] - ↑
Hashimoto, K., Sawa, A., & Iyo, M. (2007).
Increased levels of glutamate in brains from patients with mood disorders. Biological psychiatry, 62(11), 1310-6. [PubMed:17574216] [WorldCat] [DOI] - ↑
Mitani, H., Shirayama, Y., Yamada, T., Maeda, K., Ashby, C.R., & Kawahara, R. (2006).
Correlation between plasma levels of glutamate, alanine and serine with severity of depression. Progress in neuro-psychopharmacology & biological psychiatry, 30(6), 1155-8. [PubMed:16707201] [WorldCat] [DOI] - ↑
Choudary, P.V., Molnar, M., Evans, S.J., Tomita, H., Li, J.Z., Vawter, M.P., ..., & Jones, E.G. (2005).
Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proceedings of the National Academy of Sciences of the United States of America, 102(43), 15653-8. [PubMed:16230605] [PMC] [WorldCat] [DOI] - ↑
Mineur, Y.S., Picciotto, M.R., & Sanacora, G. (2007).
Antidepressant-like effects of ceftriaxone in male C57BL/6J mice. Biological psychiatry, 61(2), 250-2. [PubMed:16860779] [WorldCat] [DOI] - ↑
Sanacora, G., Kendell, S.F., Levin, Y., Simen, A.A., Fenton, L.R., Coric, V., & Krystal, J.H. (2007).
Preliminary evidence of riluzole efficacy in antidepressant-treated patients with residual depressive symptoms. Biological psychiatry, 61(6), 822-5. [PubMed:17141740] [PMC] [WorldCat] [DOI] - ↑
Cui, W., Mizukami, H., Yanagisawa, M., Aida, T., Nomura, M., Isomura, Y., ..., & Aizawa, H. (2014).
Glial dysfunction in the mouse habenula causes depressive-like behaviors and sleep disturbance. The Journal of neuroscience : the official journal of the Society for Neuroscience, 34(49), 16273-85. [PubMed:25471567] [PMC] [WorldCat] [DOI] - ↑
Gigante, A.D., Bond, D.J., Lafer, B., Lam, R.W., Young, L.T., & Yatham, L.N. (2012).
Brain glutamate levels measured by magnetic resonance spectroscopy in patients with bipolar disorder: a meta-analysis. Bipolar disorders, 14(5), 478-87. [PubMed:22834460] [WorldCat] [DOI] - ↑
Fiorentino, A., Sharp, S.I., & McQuillin, A. (2015).
Association of rare variation in the glutamate receptor gene SLC1A2 with susceptibility to bipolar disorder and schizophrenia. European journal of human genetics : EJHG, 23(9), 1200-6. [PubMed:25406999] [PMC] [WorldCat] [DOI] - ↑
Serra, G., Demontis, F., Serra, F., De Chiara, L., Spoto, A., Girardi, P., ..., & Serra, G. (2014).
Memantine: New prospective in bipolar disorder treatment. World journal of psychiatry, 4(4), 80-90. [PubMed:25540723] [PMC] [WorldCat] [DOI] - ↑ 53.0 53.1
Ting, J.T., & Feng, G. (2008).
Glutamatergic Synaptic Dysfunction and Obsessive-Compulsive Disorder. Current chemical genomics, 2, 62-75. [PubMed:19768139] [PMC] [WorldCat] [DOI] - ↑
Mattheisen, M., Samuels, J.F., Wang, Y., Greenberg, B.D., Fyer, A.J., McCracken, J.T., ..., & Nestadt, G. (2015).
Genome-wide association study in obsessive-compulsive disorder: results from the OCGAS. Molecular psychiatry, 20(3), 337-44. [PubMed:24821223] [PMC] [WorldCat] [DOI] - ↑
Autism Genome Project Consortium, Szatmari, P., Paterson, A.D., Zwaigenbaum, L., Roberts, W., Brian, J., ..., & Meyer, K.J. (2007).
Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nature genetics, 39(3), 319-28. [PubMed:17322880] [PMC] [WorldCat] [DOI] - ↑
Aida, T., Yoshida, J., Nomura, M., Tanimura, A., Iino, Y., Soma, M., ..., & Tanaka, K. (2015).
Astroglial glutamate transporter deficiency increases synaptic excitability and leads to pathological repetitive behaviors in mice. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology, 40(7), 1569-79. [PubMed:25662838] [PMC] [WorldCat] [DOI] - ↑ 57.0 57.1
Jen, J.C., Wan, J., Palos, T.P., Howard, B.D., & Baloh, R.W. (2005).
Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology, 65(4), 529-34. [PubMed:16116111] [WorldCat] [DOI] - ↑ 58.0 58.1
de Vries, B., Mamsa, H., Stam, A.H., Wan, J., Bakker, S.L., Vanmolkot, K.R., ..., & van den Maagdenberg, A.M. (2009).
Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake. Archives of neurology, 66(1), 97-101. [PubMed:19139306] [WorldCat] [DOI] - ↑
Watanabe, T., Morimoto, K., Hirao, T., Suwaki, H., Watase, K., & Tanaka, K. (1999).
Amygdala-kindled and pentylenetetrazole-induced seizures in glutamate transporter GLAST-deficient mice. Brain research, 845(1), 92-6. [PubMed:10529447] [WorldCat] [DOI] - ↑
Kong, Q., Takahashi, K., Schulte, D., Stouffer, N., Lin, Y., & Lin, C.L. (2012).
Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiology of disease, 47(2), 145-54. [PubMed:22513140] [PMC] [WorldCat] [DOI] - ↑ 61.0 61.1
Kong, Q., Chang, L.C., Takahashi, K., Liu, Q., Schulte, D.A., Lai, L., ..., & Lin, C.L. (2014).
Small-molecule activator of glutamate transporter EAAT2 translation provides neuroprotection. The Journal of clinical investigation, 124(3), 1255-67. [PubMed:24569372] [PMC] [WorldCat] [DOI] - ↑
Nakagawa, T., & Kaneko, S. (2013).
SLC1 glutamate transporters and diseases: psychiatric diseases and pathological pain. Current molecular pharmacology, 6(2), 66-73. [PubMed:23876150] [WorldCat] - ↑
Takahashi, K., Foster, J.B., & Lin, C.L. (2015).
Glutamate transporter EAAT2: regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cellular and molecular life sciences : CMLS, 72(18), 3489-506. [PubMed:26033496] [WorldCat] [DOI] - ↑
Shen, H.W., Scofield, M.D., Boger, H., Hensley, M., & Kalivas, P.W. (2014).
Synaptic glutamate spillover due to impaired glutamate uptake mediates heroin relapse. The Journal of neuroscience : the official journal of the Society for Neuroscience, 34(16), 5649-57. [PubMed:24741055] [PMC] [WorldCat] [DOI] - ↑
Chen, K.H., Reese, E.A., Kim, H.W., Rapoport, S.I., & Rao, J.S. (2011).
Disturbed neurotransmitter transporter expression in Alzheimer's disease brain. Journal of Alzheimer's disease : JAD, 26(4), 755-66. [PubMed:21743130] [PMC] [WorldCat] [DOI] - ↑
Mookherjee, P., Green, P.S., Watson, G.S., Marques, M.A., Tanaka, K., Meeker, K.D., ..., & Cook, D.G. (2011).
GLT-1 loss accelerates cognitive deficit onset in an Alzheimer's disease animal model. Journal of Alzheimer's disease : JAD, 26(3), 447-55. [PubMed:21677376] [PMC] [WorldCat] [DOI] - ↑
Scimemi, A., Meabon, J.S., Woltjer, R.L., Sullivan, J.M., Diamond, J.S., & Cook, D.G. (2013).
Amyloid-β1-42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. The Journal of neuroscience : the official journal of the Society for Neuroscience, 33(12), 5312-8. [PubMed:23516295] [PMC] [WorldCat] [DOI] - ↑
Zumkehr, J., Rodriguez-Ortiz, C.J., Cheng, D., Kieu, Z., Wai, T., Hawkins, C., ..., & Kitazawa, M. (2015).
Ceftriaxone ameliorates tau pathology and cognitive decline via restoration of glial glutamate transporter in a mouse model of Alzheimer's disease. Neurobiology of aging, 36(7), 2260-2271. [PubMed:25964214] [WorldCat] [DOI] - ↑
Takahashi, K., Kong, Q., Lin, Y., Stouffer, N., Schulte, D.A., Lai, L., ..., & Lin, C.L. (2015).
Restored glial glutamate transporter EAAT2 function as a potential therapeutic approach for Alzheimer's disease. The Journal of experimental medicine, 212(3), 319-32. [PubMed:25711212] [PMC] [WorldCat] [DOI] - ↑
Spreux-Varoquaux, O., Bensimon, G., Lacomblez, L., Salachas, F., Pradat, P.F., Le Forestier, N., ..., & Meininger, V. (2002).
Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: a reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. Journal of the neurological sciences, 193(2), 73-8. [PubMed:11790386] [WorldCat] [DOI] - ↑
Rothstein, J.D., Martin, L.J., & Kuncl, R.W. (1992).
Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. The New England journal of medicine, 326(22), 1464-8. [PubMed:1349424] [WorldCat] [DOI] - ↑
Rothstein, J.D., Van Kammen, M., Levey, A.I., Martin, L.J., & Kuncl, R.W. (1995).
Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Annals of neurology, 38(1), 73-84. [PubMed:7611729] [WorldCat] [DOI] - ↑
Howland, D.S., Liu, J., She, Y., Goad, B., Maragakis, N.J., Kim, B., ..., & Rothstein, J.D. (2002).
Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proceedings of the National Academy of Sciences of the United States of America, 99(3), 1604-9. [PubMed:11818550] [PMC] [WorldCat] [DOI] - ↑
Tong, J., Huang, C., Bi, F., Wu, Q., Huang, B., Liu, X., ..., & Xia, X.G. (2013).
Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. The EMBO journal, 32(13), 1917-26. [PubMed:23714777] [PMC] [WorldCat] [DOI] - ↑
Rothstein, J.D., Patel, S., Regan, M.R., Haenggeli, C., Huang, Y.H., Bergles, D.E., ..., & Fisher, P.B. (2005).
Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature, 433(7021), 73-7. [PubMed:15635412] [WorldCat] [DOI] - ↑
Arzberger, T., Krampfl, K., Leimgruber, S., & Weindl, A. (1997).
Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington's disease--an in situ hybridization study. Journal of neuropathology and experimental neurology, 56(4), 440-54. [PubMed:9100675] [WorldCat] [DOI] - ↑ 77.0 77.1
Faideau, M., Kim, J., Cormier, K., Gilmore, R., Welch, M., Auregan, G., ..., & Bonvento, G. (2010).
In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington's disease subjects. Human molecular genetics, 19(15), 3053-67. [PubMed:20494921] [PMC] [WorldCat] [DOI] - ↑
Estrada-Sánchez, A.M., Montiel, T., Segovia, J., & Massieu, L. (2009).
Glutamate toxicity in the striatum of the R6/2 Huntington's disease transgenic mice is age-dependent and correlates with decreased levels of glutamate transporters. Neurobiology of disease, 34(1), 78-86. [PubMed:19168136] [WorldCat] [DOI] - ↑
Miller, B.R., Dorner, J.L., Shou, M., Sari, Y., Barton, S.J., Sengelaub, D.R., ..., & Rebec, G.V. (2008).
Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington's disease phenotype in the R6/2 mouse. Neuroscience, 153(1), 329-37. [PubMed:18353560] [PMC] [WorldCat] [DOI] - ↑ Freilinger T, Koch J, Dichgans M, Mamsa H, Jen JC.
A novel mutation in slc1a3 associated with pure hemiplegic migraine.
J Headache Pain 11(Suppl 1): S90, 2010. - ↑
De Fusco, M., Marconi, R., Silvestri, L., Atorino, L., Rampoldi, L., Morgante, L., ..., & Casari, G. (2003).
Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nature genetics, 33(2), 192-6. [PubMed:12539047] [WorldCat] [DOI] - ↑
Suzuki, M., Van Paesschen, W., Stalmans, I., Horita, S., Yamada, H., Bergmans, B.A., ..., & Seki, G. (2010).
Defective membrane expression of the Na(+)-HCO(3)(-) cotransporter NBCe1 is associated with familial migraine. Proceedings of the National Academy of Sciences of the United States of America, 107(36), 15963-8. [PubMed:20798035] [PMC] [WorldCat] [DOI] - ↑
Anttila, V., Stefansson, H., Kallela, M., Todt, U., Terwindt, G.M., Calafato, M.S., ..., & International Headache Genetics Consortium (2010).
Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nature genetics, 42(10), 869-73. [PubMed:20802479] [PMC] [WorldCat] [DOI] - ↑
Srinivasan, R., Sailasuta, N., Hurd, R., Nelson, S., & Pelletier, D. (2005).
Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain : a journal of neurology, 128(Pt 5), 1016-25. [PubMed:15758036] [WorldCat] [DOI] - ↑
Tisell, A., Leinhard, O.D., Warntjes, J.B., Aalto, A., Smedby, Ö., Landtblom, A.M., & Lundberg, P. (2013).
Increased concentrations of glutamate and glutamine in normal-appearing white matter of patients with multiple sclerosis and normal MR imaging brain scans. PloS one, 8(4), e61817. [PubMed:23613944] [PMC] [WorldCat] [DOI] - ↑
Stover, J.F., Pleines, U.E., Morganti-Kossmann, M.C., Kossmann, T., Lowitzsch, K., & Kempski, O.S. (1997).
Neurotransmitters in cerebrospinal fluid reflect pathological activity. European journal of clinical investigation, 27(12), 1038-43. [PubMed:9466133] [WorldCat] [DOI] - ↑
Vercellino, M., Merola, A., Piacentino, C., Votta, B., Capello, E., Mancardi, G.L., ..., & Cavalla, P. (2007).
Altered glutamate reuptake in relapsing-remitting and secondary progressive multiple sclerosis cortex: correlation with microglia infiltration, demyelination, and neuronal and synaptic damage. Journal of neuropathology and experimental neurology, 66(8), 732-9. [PubMed:17882017] [WorldCat] [DOI] - ↑
Smith, T., Groom, A., Zhu, B., & Turski, L. (2000).
Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nature medicine, 6(1), 62-6. [PubMed:10613825] [WorldCat] [DOI] - ↑
Pitt, D., Werner, P., & Raine, C.S. (2000).
Glutamate excitotoxicity in a model of multiple sclerosis. Nature medicine, 6(1), 67-70. [PubMed:10613826] [WorldCat] [DOI] - ↑
Thier, S., Lorenz, D., Nothnagel, M., Poremba, C., Papengut, F., Appenzeller, S., ..., & Kuhlenbäumer, G. (2012).
Polymorphisms in the glial glutamate transporter SLC1A2 are associated with essential tremor. Neurology, 79(3), 243-8. [PubMed:22764253] [PMC] [WorldCat] [DOI] - ↑
Lee, M., Cheng, M.M., Lin, C.Y., Louis, E.D., Faust, P.L., & Kuo, S.H. (2014).
Decreased EAAT2 protein expression in the essential tremor cerebellar cortex. Acta neuropathologica communications, 2, 157. [PubMed:25391854] [PMC] [WorldCat] [DOI] - ↑
Mallolas, J., Hurtado, O., Castellanos, M., Blanco, M., Sobrino, T., Serena, J., ..., & Dávalos, A. (2006).
A polymorphism in the EAAT2 promoter is associated with higher glutamate concentrations and higher frequency of progressing stroke. The Journal of experimental medicine, 203(3), 711-7. [PubMed:16520390] [PMC] [WorldCat] [DOI] - ↑
Mitani, A., & Tanaka, K. (2003).
Functional changes of glial glutamate transporter GLT-1 during ischemia: an in vivo study in the hippocampal CA1 of normal mice and mutant mice lacking GLT-1. The Journal of neuroscience : the official journal of the Society for Neuroscience, 23(18), 7176-82. [PubMed:12904478] [PMC] [WorldCat] - ↑
Inui, T., Alessandri, B., Heimann, A., Nishimura, F., Frauenknecht, K., Sommer, C., & Kempski, O. (2013).
Neuroprotective effect of ceftriaxone on the penumbra in a rat venous ischemia model. Neuroscience, 242, 1-10. [PubMed:23523747] [WorldCat] [DOI] - ↑
Hu, Y.Y., Xu, J., Zhang, M., Wang, D., Li, L., & Li, W.B. (2015).
Ceftriaxone modulates uptake activity of glial glutamate transporter-1 against global brain ischemia in rats. Journal of neurochemistry, 132(2), 194-205. [PubMed:25270764] [WorldCat] [DOI] - ↑
Tao, Y.X., Gu, J., & Stephens, R.L. (2005).
Role of spinal cord glutamate transporter during normal sensory transmission and pathological pain states. Molecular pain, 1, 30. [PubMed:16242033] [PMC] [WorldCat] [DOI] - ↑
Hu, Y., Li, W., Lu, L., Cai, J., Xian, X., Zhang, M., ..., & Li, L. (2010).
An anti-nociceptive role for ceftriaxone in chronic neuropathic pain in rats. Pain, 148(2), 284-301. [PubMed:20022427] [WorldCat] [DOI] - ↑
Gegelashvili, G., & Bjerrum, O.J. (2014).
High-affinity glutamate transporters in chronic pain: an emerging therapeutic target. Journal of neurochemistry, 131(6), 712-30. [PubMed:25270665] [WorldCat] [DOI] - ↑
Harada, T., Harada, C., Nakamura, K., Quah, H.M., Okumura, A., Namekata, K., ..., & Tanaka, K. (2007).
The potential role of glutamate transporters in the pathogenesis of normal tension glaucoma. The Journal of clinical investigation, 117(7), 1763-70. [PubMed:17607354] [PMC] [WorldCat] [DOI] - ↑
Yanagisawa, M., Aida, T., Takeda, T., Namekata, K., Harada, T., Shinagawa, R., & Tanaka, K. (2015).
Arundic acid attenuates retinal ganglion cell death by increasing glutamate/aspartate transporter expression in a model of normal tension glaucoma. Cell death & disease, 6, e1693. [PubMed:25789968] [PMC] [WorldCat] [DOI]